

Журнал физической химии, 2019, T. 93, № 4, стр. 565-572
Механизм сорбционного взаимодействия L-аланина с углеродными нанотрубками
Е. В. Бутырская a, *, С. А. Запрягаев a, Е. А. Измайлова a, В. Ф. Селеменев a
a Воронежский государственный университет
Воронеж, Россия
* E-mail: bev5105@yandex.ru
Поступила в редакцию 21.05.2018
Аннотация
В рамках теории функционала плотности с учетом дисперсионных поправок выполнено квантово-химическое моделирование комплекса, состоящего из L-аланина и модельной углеродной нанотрубки при расположении аминокислоты на наружной боковой поверхности, внутри и на конце трубки. Проведен анализ механизма взаимодействия аминокислоты и нанотрубки и оценена энергия ван-дер-ваальсовых взаимодействий сорбент–сорбат. Построены и проинтерпретированы изотермы адсорбции L-аланина на многостенных углеродных нанотрубках MKN-MWCNT-P5000 (Канада) из водного раствора.
Углеродные нанотрубки (УНТ) представляют собой новый класс наноматериалов, имеющих огромный потенциал для разнообразных технологических приложений [1–9]. Наиболее значимыми направлениями применения УНТ являются биомедицина, материаловедение и наноэлектроника.
Понимание природы взаимодействия УНТ с аминокислотами (АК) имеет большое значение для различных биомедицинских приложений. Исследования взаимодействий АК с УНТ выполняются в основном методами квантовой химии, молекулярной динамики, а также физико-химическими методами [10–16]. Результаты исследований указывают на возможность адсорбции АК боковой поверхностью УНТ [14], на высокую вероятность втягивания АК внутрь УНТ, возможность образования внутри УНТ нанопроволок и наноспиралей АК, взаимодействующих с внутренней поверхностью нанотрубки [15, 16], высокую энергию адсорбции открытыми концами и дефектами боковой поверхности УНТ. Несмотря на большое число исследований взаимодействий АК и УНТ в литературе практически не представлены результаты квантово-химических расчетов с учетом дисперсионных поправок, что приводит к большим ошибкам в величине энергии адсорбции вследствие большой поляризуемости углеродных наночастиц. Очень немногочисленны работы, в которых построены и исследованы изотермы адсорбции АК на УНТ, недостаточно представлен анализ механизма взаимодействия УНТ с аминокислотами. В нашей работе [17] исследована адсорбция L- и D-аланина на одностенных УНТ MKN-SWCNT S1. Установленное в [17] большое сродство УНТ к D-аланину может служить основой энантиоразделения аминокислот на УНТ.
Цель настоящей работы – квантово-химическое моделирование адсорбции аланина на модельных углеродных нанотрубках и анализ механизма их взаимодействия, построение и интерпретация изотермы адсорбции L-аланина на многостенных углеродных нанотрубках из водного раствора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Построение изотерм. Для построения изотерм адсорбции использовали многостенные углеродные нанотрубки MKN-MWCNT-P5000 (Канада). В качестве аминокислот использовали α-аланин (L) от производителя Sigma Aldrich (Russia). Углеродные нанотрубки заливали водным раствором аланина (pH ∼ 7). Полученные суспензии подвергали воздействию ультразвука в течение 3 мин с использованием ультразвуковой установки МЭФ91. Затем суспензию непрерывно перемешивали в течение 4 ч в шейкер-инкубаторе ES-20, после чего выдерживали 24 ч в статических условиях при температуре 20°С для установления равновесия. Данный режим подготовки выявлен в результате кинетических исследований. Через 24 ч суспензию центрифугировали и определяли концентрацию аминокислоты в супернатанте методом спектрофотометрии.
Компьютерное моделирование. Проводили оптимизацию структуры цвиттер-иона аланина, углеродной нанторубки и системы аланин + УНТ в водном растворе. В качестве УНТ выбрали нанотрубку хиральности (6.6) (диаметр 8.1 Å) с длиной 13.53 Å. В стартовых структурах сорбат + УНТ цвиттер-ионы аланина располагали тремя способами: на внешней боковой стороне, на открытом конце и внутри УНТ. Оптимизацию структур и расчет энергий проводили с использованием программы Gaussian 09 [18, 19] методом B3LYP/6-31G(d,p) с учетом дисперсионной поправки GD3 [20]. Учет растворителя (воды) выполняли в рамках модели сольватации РСМ [21]. Расчеты проводили в Суперкомпьютерном центре Воронежского государственного университета [22].
Энергии сорбции рассчитывали по формуле:
(1)
$E_{{\text{ }}}^{c} = E_{{}}^{{{\text{Ala}}}} + E_{{}}^{{{\text{У Н Т }}}} - E_{{}}^{{{\text{Ala}} + {\text{У Н Т }}}},$Расчет энергии ван-дер-ваальсова (ВДВ) взаимодействия между аминокислотой и нанотрубкой. ВДВ-взаимодействие между двумя молекулами определяется тремя составляющими (ориентационное – обусловлено взаимодействием собственных дипольных моментов молекул, индукционное – взаимодействием собственного дипольного момента одной молекулы с индуцированным им дипольным моментом другой молекулы, и дисперсионное – взаимодействием флуктуационных “мгновенных” дипольных моментов молекул):
(2)
${{W}_{{{\text{о р и е н }}}}} = - \frac{{2d_{{{\text{А К }}}}^{2}d_{{{\text{У Н Т }}}}^{2}}}{{3kT{{r}^{6}}}},$Индукционная составляющая энергии ВДВ-взаимодействия между системой с дипольным моментом dАК и бездипольной молекулой, обладающей средней статической поляризуемостью ${{\bar {\alpha }}_{{{\text{У Н Т }}}}}$ определяется по формуле [23]
(3)
${{W}_{{{\text{и н д }}}}} = - \frac{{{{{\bar {\alpha }}}_{{{\text{У Н Т }}}}}d_{{{\text{А К }}}}^{2}}}{{{{r}^{6}}}}.$(4)
${{W}_{{{\text{д и с п }}}}} = - \frac{3}{2}\frac{{{{I}_{{{\text{А К }}}}}{{I}_{{{\text{У Н Т }}}}}}}{{{{I}_{{{\text{А К }}}}} + {{I}_{{{\text{У Н Т }}}}}}}\frac{{{{\alpha }_{{{\text{А К }}}}}{{{\bar {\alpha }}}_{{{\text{У Н Т }}}}}}}{{{{r}^{6}}}},$ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Компьютерный эксперимент
Оптимизированные структуры систем цвиттер-ион аланина + УНТ для трех случаев взаимного расположения аминокислоты (на боковой стороне, на конце и внутри УНТ) в водном растворе представлены на рис. 1–3). Тип адсорбции аминокислоты на УНТ (физическая, химическая) может быть выявлен анализом оптимизированных структур, что представлено ниже для различных случаев взаимного расположения сорбент–сорбат.
Рис. 1.
Оптимизированная структура системы цвиттер-иона L-аланина при расположении аминокислоты на боковой поверхности нанотрубки; Еадс= 9.16 ккал/моль, О(1)СУНТ = 3.29 Å, О(2)СУНТ = 3.35 Å, NCУНТ= 3.47 Å, $\sum {{{q}_{{{\text{Ala}}}}}} = - 0.011$, $\sum {{{q}_{{{\text{У Н Т }}}}}} = 0.011$.
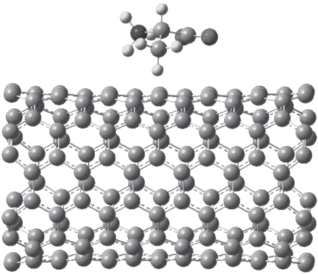
Рис. 2.
Оптимизированная структура системы цвиттер-ион аланина внутри углеродной нанотрубки; Eадс= = 22.59 ккал/моль, О(1)СУНТ = 3.45 Å, О(2)СУНТ = 3.49 Å, NCУНТ = 3.52 Å, ${{{\text{O}}}_{{(1)}}}{\text{C}}_{{{\text{У Н Т }}}}^{{{\text{end}}}} = 6.77$ Å, ${{{\text{O}}}_{{(2)}}}{\text{C}}_{{{\text{У Н Т }}}}^{{{\text{end}}}} = 6.67$ Å, ${\text{NC}}_{{{\text{У Н Т }}}}^{{{\text{end}}}} = 6.67$ Å, $\sum {{{q}_{{{\text{Ala}}}}}} = - 0.037$, $\sum {{{q}_{{{\text{У Н Т }}}}}} = - 0.037$.
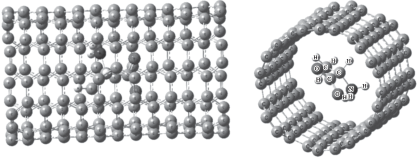
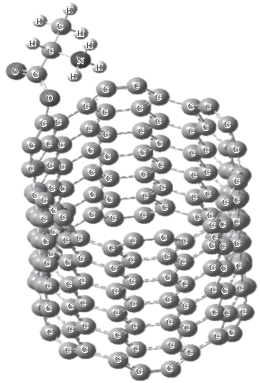
Рис. 3.
Оптимизированная структура системы цвиттер-иона L-аланина при расположении аминокислоты на конце нанотрубки диаметра 8.1 Å; Еадс= 31.38 ккал/моль; О(1)СУНТ = 1.45 Å, О(2)СУНТ = 2.81 Å, NCУНТ = 3.34 Å, $\sum {{{q}_{{{\text{Ala}}}}}} = 0.503$, $\sum {{{q}_{{{\text{У Н Т }}}}}} = - 0.503$ (диссоциативная хемосорбция на короткой).
Случай 1. Аминокислота расположена на боковой поверхности УНТ. На рис. 1 представлена оптимизированная структура исследуемой системы. Расположение АК относительно УНТ на рис. 1 охарактеризовано расстояниями от атомов N и O аминокислоты до ближайших атомов углерода УНТ. Все расстояния от атомов аланина до атомов С нанотрубки значительно меньше сумм соответствующих ковалентных радиусов, поэтому ковалентные связи между УНТ и аминокислотой отсутствуют. Свойства оптимизированного комплекса (рис. 1) представлены энергией адсорбции аланина на УНТ (Еадс), рассчитанной по формуле (1); суммой зарядов по Малликену на атомах аланина – $\sum {{{q}_{{{\text{Ala}}}}}} $ и атомах УНТ – $\sum {{{q}_{{{\text{У Н Т }}}}}} $, рассчитанных по программе GAUSSIAN. Незначительный перенос электронной плотности с УНТ на цвиттер-ион позволяет сделать вывод об отсутствии донорно-акцепторного взаимодействия сорбент–сорбат.
Таким образом, наибольший вклад в образование комплекса дают межмолекулярные силы Ван-дер-Ваальса (ВДВ), оценка которых приведена ниже. Для расчета ВДВ-взаимодействий в системе (рис. 1) оценим дипольный момент адсорбата. Сравнение зарядов на атомах по Малликену аминокислоты в комплексе (адсорбат, рис. 1) и в растворе (адсорбтив) показывает, что их различие очень незначительно. Поэтому дипольные момента адсорбтива и адсорбата можно считать одинаковыми, их модули равны 11.39 D (рассчитано по программе Gaussian для адсорбтива). Расчет показал, что собственный дипольный момент модельной УНТ равен нулю, поэтому ориентационное взаимодействие (2) между УНТ и аминокислотой в исследуемом комплексе (рис. 1) отсутствует.
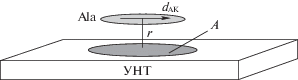
Формула (3) для индукционных взаимодействий предполагает, что электрическое поле, создаваемое диполем аминокислоты, однородно в пределах УНТ. Это условие недостаточно хорошо выполняется для исследуемой системы, поскольку трубка имеет достаточно большие размеры. Поэтому величину ${{\bar {\alpha }}_{{{\text{У Н Т }}}}}$ в (3) оценивали как произведение поляризуемости одного атома С на число атомов С в УНТ (рис. 1), которые “испытывают” влияние электрического поля диполя аминокислоты. Для этого проводили анализ зарядов на атомах по Малликену исследуемой системы (рис. 1), показавший, что при образовании комплекса происходит изменение зарядов не на всех атомах углерода УНТ. Искомая часть поверхности УНТ находится ближе к аминокислоте в небольшой окрестности ее “проекции” на поверхность трубки, что качественно изображено на рис. 4. При удалении от этой области изменение зарядов на атомах УНТ практически отсутствует (по модулю меньше 0.005).
Рис. 4.
Модель области (А) изменения зарядов по Малликену на атомах углерода нанотрубки при образовании комплекса УНТ–аланин (случай 1) в водном растворе.
Поляризуемость одного атома углерода ${{\alpha }_{{\text{C}}}}$ оценивали по формуле Клаузиуса–Моссотти:
(5)
$\frac{{\varepsilon - 1}}{{\varepsilon + 2}}\frac{M}{\rho } = \frac{{4\pi }}{3}{{N}_{{\text{A}}}}{{\alpha }_{{\text{C}}}},$Число атомов углерода в оптимизированной системе, для которых модуль изменения зарядов на атомах по Малликену, больше 0.005 равно N = 12. Умножение N на ${{\alpha }_{{\text{C}}}}$ дает оценку поляризуемости области УНТ, в которой происходит наиболее существенное изменение электронной плотности при образовании комплекса с УНТ (заштрихована на рис. 4):
(6)
${{\bar {\alpha }}_{{{\text{У Н Т }}}}} = {{\alpha }_{{\text{C}}}}N = 9.968 \times {{10}^{{ - 24}}}\;{\text{с }}{{{\text{м }}}^{3}}.$Оценку вклада дисперсионной составляющей в энергию ВДВ-взаимодействия комплекса оценим по формуле (4). При этом в качестве потенциалов ионизации АК и УНТ взяты энергии высшей занятой молекулярной орбитали с противоположным знаком (теорема Купманса). Потенциалы ионизации аминокислоты и УНТ, рассчитанные по программе Gaussian равны
Расчет по формуле (4) дает для энергии дисперсионных взаимодействий
(7)
$\begin{gathered} \left| {{{W}_{{{\text{д и с п }}}}}} \right| = 2.42\;{\text{к к а л /м о л ь }}, \\ \left| {{{W}_{{{\text{В Д В }}}}}} \right| = \left| {{{W}_{{{\text{и н д }}}}}} \right| + \left| {{{W}_{{{\text{д и с п }}}}}} \right| = 8.51\;{\text{к к а л /м о л ь }}, \\ \end{gathered} $Случай 2. Аминокислота внутри УНТ. Оптимизированная структура данной системы в водном растворе представлена на рис. 2. Расстояния от атомов кислорода и атома азота до ближайших атомов углерода нанотрубки СУНТ в рассматриваемом комплексе больше на ∼0.15 Å и 0.05 Å соответственно, чем в предыдущем случае. На рис. 2 также представлены энергия адсорбции по формуле (1), сумма зарядов на атомах аминокислоты и УНТ по Малликену, рассчитанные по программе Gaussian. Энергия адсорбции в данном случае увеличена более, чем в 2 раза по сравнению с предыдущем случаем, перенос электронной плотности с УНТ на АК, хотя и мал, но в рассматриваемом случае также в 3.4 раза выше, чем в случае 1. Более существенное изменение свойств комплекса УНТ с АК внутри УНТ обусловлено тем, что цвиттер-ион в случае втягивания в УНТ взаимодействует со всеми окружающими его стенками УНТ, а не с одной стороной нанотрубки, как в предыдущем случае.
В рассматриваемом случае так же, как в случае 1, все расстояния от атомов аланина до атомов углерода нанотрубки значительно меньше сумм соответствующих ковалентных радиусов, поэтому ковалентные связи между УНТ и аминокислотой отсутствуют. Равенство нулю дипольного момента УНТ и незначительный перенос электронной плотности с УНТ на цвиттер-ион приводят к выводу об отсутствии ориентационного и донорно-акцепторного взаимодействий между аланином и нанотрубкой и в случае 2.
Оценим энергию индукционного ВДВ-взаимодействия сорбент–сорбат для случая нахождения аминокислоты внутри УНТ. Анализ зарядов на атомах аланина внутри УНТ, как и в случае 1, показывает, что они близки к таковым на индивидуальной аминокислоте, и, таким образом, дипольный момент сорбата можно считать равным дипольному моменту сорбтива (11.39 D).
При оценке ${{W}_{{{\text{и н д }}}}}$ по формуле (3) в качестве расстояния r выбрали расстояние, равное половине диаметра УНТ (r = 4.05 Å). Это обусловлено тем, что в данном случае изменение зарядов на атомах УНТ при внедрении аминокислоты внутрь УНТ имеет место в окрестности “пояса” УНТ, окружающего аминокислоту, и центр вектора дипольного момента аминокислоты приближенно можно считать лежащим на оси УНТ. Для оценки ${{\bar {\alpha }}_{{{\text{У Н Т }}}}}$, входящей в (3), подсчитали число атомов углерода в оптимизированной системе, для которых модуль изменения зарядов на атомах по Малликену больше 0.005. При этом было получено, что для случая 2 число атомов углерода с наиболее существенным изменением зарядов на атомах по Малликену N = 49. ${{\bar {\alpha }}_{{{\text{У Н Т }}}}} = {{\alpha }_{{\text{C}}}}N = $ $ = 4.07 \times {{10}^{{ - 23}}}$ см3. Расчет по формуле (3) с данным ${{\bar {\alpha }}_{{{\text{У Н Т }}}}}$, ${{d}_{{{\text{А К }}}}} = 11.39$D (рассчитано по программе Gaussian для адсорбтива), r = 4.05 Å дает $\left| {{{W}_{{{\text{и н д }}}}}} \right| = 16.86$ ккал/моль.
Для расчета энергии дисперсионного взаимодействия используем формулу (4). Так как ${{I}_{{{\text{А К }}}}}\, = \,0.231$ а.е.е., ${{I}_{{{\text{У Н Т }}}}} = 0.175$ а.е.е., ${{\alpha }_{{{\text{А К }}}}} = 7.925 \times $ $ \times \;{{10}^{{ - 24}}}$ см3, (см. выше), ${{\bar {\alpha }}_{{{\text{У Н Т }}}}} = 4.07 \times {{10}^{{ - 23}}}$ см3, $r = 4.05$ Å, определенные для рассматриваемого случая, получаем
тогдаВзаимодействие АК с УНТ, несмотря на значительную величину энергии взаимодействия, при расположении аминокислоты внутри УНТ является ван-дер-ваальсовым, причем вклад в Еадс индукционных взаимодействий составляет 71.5%, дисперионных взаимодействий – 28.5%. Бóльшие, по сравнению с предыдущим случаем, абсолютные значения Еадс, ${{W}_{{{\text{и н д }}}}}$ и ${{W}_{{{\text{д и с п }}}}}$ обусловлены тем, что внутри УНТ аминокислота взаимодействует со всеми стенками трубки, а не с одной стороной поверхности как в предыдущем случае. Данный вывод также подтверждается результатами молекулярно-динамического моделирования [24].
Случай 3. Аминокислота на открытом конце УНТ. Оптимизированная структура данной системы представлена на рис. 3. В данном случае между атомом кислорода аланина и атомом углерода УНТ образуется ковалентная связь с длиной 1.45 Å (данная величина немного меньше 1.48 Å – суммы ковалентных радиусов атомов О и С, и характерна для длин одинарных С–О-связей в органических соединениях). Анализ зарядов на атомах АК и УНТ (рис. 3) позволяет сделать вывод о достаточно большом переносе электронной плотности с аминокислоты на УНТ. При этом наибольшее изменение зарядов на атомах С нанотрубки происходит на полуповерхности УНТ, в которую входит атом СУНТ = С*, участвующий в ковалентной связи с аминокислотой. При этом изменение зарядов в первом слое нанотрубки, включающем атом С*, хотя и наибольшее, однако составляет всего 22.2% от величины $\sum {{{q}_{{{\text{У Н Т }}}}}} = - 0.503$. Это обусловлено тем, что в данном случае, изменение зарядов на атомах С нанотрубки, расположенных вблизи АК, существенно больше, чем для двух предыдущих случаев и передается всей сопряженной системе, хотя и уменьшается при приближении к свободному концу УНТ. Модуль зарядов на атомах углерода трубки осциллирует при удалении от С* по образующей трубке к противоположному концу УНТ с убывающей огибающей. Значительный перенос электронной плотности с цвиттер-иона аланина на УНТ указывает на то, что в данном случае помимо ковалентного связывания имеет место образование комплекса с переносом заряда.
Таким образом, кроме энергии ковалентного связывания, вклад в энергию адсорбции дает донорно-акцепторное взаимодействие аминокислота – УНТ, а также ВДВ-взаимодействия. Оценка ВДВ-взаимодействий затруднительна, поскольку образование ковалентной связи аланин–УНТ приводит к существенному изменению зарядов на атомах как аминокислоты, так и УНТ по сравнению с индивидуальными сорбатом и сорбентом. Следовательно, дипольный момент адсорбата отличается от дипольного момента адсорбтива и последний не может быть использован при оценке ${{W}_{{{\text{В Д В }}}}}$ по формулам (3) и (4). Анализ зарядов на атомах по Малликену УНТ в комплексе (рис. 2) показывает, что изменение электронной плотности вблизи атома С* по цепи сопряжения передается почти всем атомам углерода УНТ. Следовательно, нанотрубка не может считаться точечным объектом, вследствие чего формулы (3) и (4) не являются корректными. Поэтому мы приводим лишь оценку энергии донорно-акцепторного взаимодействия, используя для ее расчета закон Кулона
(8)
$\begin{gathered} {{E}_{{{\text{д }}{\text{.а }}}}} = \frac{{{{q}_{{{\text{А К }}}}}{{q}_{{{\text{У Н Т }}}}}}}{r} = \\ = \frac{{\sum {{{q}_{{{\text{Ala}}}}}\sum {{{q}_{{{\text{У Н Т }}}}}} } }}{r}\;({\text{с и с т е м а }}\;{\text{CGSE}}), \\ \end{gathered} $При этом в качестве r = 11 Å выбрано расстояние между центрами масс УНТ и аминокислоты в комплексе (рис. 2). Расчет по формуле (8) дает
что составляет ∼23.8% энергии взаимодействия системы аланин–УНТ, рассчитанной квантово-химически (рис. 3).ИНТЕРПРЕТАЦИЯ ИЗОТЕРМ АДСОРБЦИИ
Изотерма адсорбции аланина на нанотрубках MKN-MWCNT-P5000 из водного раствора представлена на рис. 5. Изотерма имеет перегиб и два плато. Наличие второго плато может быть обусловлено полимолекулярной сорбцией или наличием энергетически неравноценных адсорбционных центров.
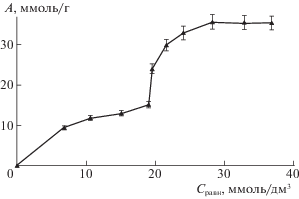
При полимолекулярной сорбции на однородной поверхности изотерма может быть описана уравнением БЭТ для жидких сред [25]:
Однако квантово-химический расчет показал также наличие энергетически неравноценных адсорбционных центров даже на модельных бездеффектных УНТ (рис. 1–3). Поэтому перегиб на изотерме может быть также обусловлен переходом от сорбции на одних центрах к сорбции на других центрах при мономолекулярной сорбции. Для исследования возможности реализации такого процесса проанализируем имеющиеся литературные данные о структурных и сорбционных свойствах УНТ.
Известно, что большая часть методов получения нанотрубок приводит к тому, что их концы обычно закрыты фуллереновыми “шапочками” (caps) [26], а боковая поверхность УНТ может иметь различные деффекты, например, моновакансии, деффекты Stone–Wales и др. [27]. При построении изотерм сорбции процедуру открытия концов УНТ не проводили, поэтому большинство использованных УНТ имеют концы, закрытые фуллереновой “шапочкой”. Как показал расчет [28], энергия взаимодействия аминокислот с фуллереном С60 в водной среде значительно меньше, чем с открытым концом УНТ и близка к энергии адсорбции боковой поверхностью УНТ. В силу этого вероятность сорбции на закрытых фуллереновыми шапками концах УНТ меньше, чем на конечных участках открытых УНТ и близка к таковой на боковой поверхности.
При проведении расчета рассматривали только бездеффектные УНТ с открытыми концами. Поэтому выполненный квантово-химический расчет лишь приближенно описывает реальный процесс сорбции и приводится для демонстрации существования различных механизмов мономолекулярной сорбции.
Незначительная концентрация моновакансий, открытых концов, и деффектов Stone–Wales нанотрубок [27] и большое аспектное число (отношение длины УНТ к диаметру ∼400), позволяют предположить, что данными участками будет сорбироваться малая часть сорбата. В результате их влияние не будет отражаться на виде изотермы, несмотря на то, что энергии сорбции на них наиболее значительны. Однако, как следует из литературных данных, значительная часть аминокислоты может войти внутрь УНТ через имеющиеся деффекты [15, 16, 24]. Поэтому, на взгляд авторов настоящей работы, наибольший вклад в мономолекулярную адсорбцию дают два механизма: 1) адсорбция аминокислоты боковой поверхностью УНТ, 2) втягивание аминокислоты внутрь УНТ через имеющиеся дефекты и открытые концы УНТ.
Известно, что в случае нескольких механизмов адсорбции, если они реализуются одновременно, изотерма представляет собой сумму изотерм, каждая из которых отвечает отдельному механизму. Так, в случае двух одновременно реализуемых ленгмюровских механизмов результирующая изотерма является так называемой изотермой биленгмюра. Уравнение изотермы биленгмюра представляет сумму двух изотерм Ленгмюра и имеет вид [29 ]
(9)
$A(C) = {{A}_{{\infty 1}}}\frac{{{{K}_{{1L}}}C}}{{1 + {{K}_{{1L}}}C}} + {{A}_{{\infty 2}}}\frac{{{{K}_{{2L}}}C}}{{1 + {{K}_{{2L}}}C}},$Молекулы аминокислоты распределены равномерно вокруг УНТ. Поэтому наиболее вероятно, что адсорбция аминокислоты боковой поверхностью и втягивание внутрь УНТ начинаются одновременно, несмотря на различие в энергиях адсорбции. В целом, по-видимому, адсорбционные места на наружной поверхности трубки заполняются быстрее вследствие их более высокой доступности, чем таковые внутри УНТ. Однако, поскольку энергия взаимодействия АК–УНТ при расположении АК внутри УНТ больше, чем таковая при адсорбции боковой поверхностью (рис. 1, 2), то в случае, когда АК одновременно находится и вблизи боковой поверхности, и вблизи дефекта, позволяющего войти внутрь нанотрубки, молекулы сорбата будут втягиваться внутрь УНТ. Отсюда можно предложить один из достаточно вероятных механизмов закрепления АК на УНТ: адсорбция боковой поверхностью и втягивание внутрь нанотрубки начинаются и заканчиваются одновременно (втягивание последних молекул сорбата внутрь и их адсорбция боковой поверхностью вблизи дефектов происходят в одно и то же время вследствие того, что вблизи дефектов находится незначительная часть поверхности трубки). В рамках такой модели первый участок изотермы, включающий первое плато, соответствует сумме вкладов двух механизмов, обусловленных адсорбционными центрами на боковой наружной и внутренней поверхностях УНТ. Участок же изотермы со вторым плато при такой интерпретации обусловлен полимолекулярной сорбцией и, при благоприятном геометрическом факторе, объемным заполнением внутренней части нанотрубки при занятых поверхностных местах. Однако, в этом случае определить и проинтерпретировать параметры модели достаточно сложно.
ЗАКЛЮЧЕНИЕ
Энергии адсорбции аланина на УНТ с открытыми концами удовлетворяют согласно результатам квантово-химического расчета условию: Eкон > Eвнутри > Eбок. При расположении АК на боковой поверхности и внутри нанотрубки закрепление сорбата на УНТ осуществляется посредством индукционных и дисперсионных взаимодействий Ван-дер-Ваальса. При этом вклад индукционных взаимодействий в 2.5 раза больше, чем дисперсионных. Соотношение Eвнутри > Eбок обусловлено тем, что внутри УНТ аминокислота взаимодействует со всеми стенками трубки, а не с одной стороной поверхности при расположении на боковой поверхности. При расположении АК на конце УНТ имеет место образование комплекса с переносом заряда и ковалентная связь сорбент–сорбат. Построенные изотермы адсорбции аланина на УНТ MKN-MWCNT-P5000 имеют перегиб и два плато. Найдены сорбционные параметры изотермы в рамках модели БЭТ. На основании анализа изотерм, результатов квантово-химического расчета и литературных данных предложена возможная модель мономолекулярной сорбции АК на УНТ со сменой механизма сорбции. Согласно этой модели, участок изотермы с первым плато отражает сумму вкладов сорбции аминокислоты боковой наружной и внутренней поверхностями УНТ, участок со вторым плато обусловлен полимолекулярной сорбцией и, при благоприятном геометрическом факторе, объемным заполнением внутренней части нанотрубки при занятых поверхностных местах.
Работа выполнена при поддержке Минобрнауки России в рамках выполнения государственного задания вузам в сфере научной деятельности на 2017–2019 годы (проект № 1.4539.2017/8.9.).
Список литературы
Раков Э.Г. // Успехи химии. 2013. Т. 82. № 1. С. 27.
Avishek S., Chengmin J., Angel A., Martí J. // Carbon. 2014. V. 79. P. 1.
Tasis D., Tagmatarchis N., Bianco A. // Chem. Rev. 2006. V. 106. P. 1105.
Yu L., Shearer C., Shapter J. // Chem. Rev. 2016. V. 116. P. 13413.
Liné C., Larue C., Flahaut E. // Carbon. 2017. V. 123. P. 767.
Brownlie L., Shapter J. // Ibid. 2018. V. 126. P. 257.
Sun L., Wang X., Wang Y., Zhang Q. // Ibid. 2017. V. 122. P. 462.
Vardharajula S., Ali S.Z., Tiwari P.M., Eroglu E. // J. Nanomedicine. 2012. V. 7. P. 5361.
Zardini H.Z., Amiri A., Shanbedi M., Maghrebi M. // Biointerfaces. 2012. V. 92. P. 196.
Leon A., Jalbout A.F., Basiuk V.A. // Chem. Phys. Let. 2008. V. 457. P. 185.
Vardanega D., Picaud F. // J. Biotechnology. 2009. V. 144. P. 96.
Ganji M.D. Density Functional Theory Based Treatment of Amino Acids Adsorption on Single-Walled Carbon Nanotubes: Diamond & Related Materials. 2009. P. 662.
Roman T., Dino W.A., Nakanishi H., Kasai H. // J. Phys. 2006. V. 38. P. 117.
Zhou J., He Z. // Carbon. 2014. V. 78. P. 500.
Garalleh H.A.L., Thamwattana N., Cox B.J., James M.H. // J. Biomaterials and Tissue Engineering. 2016. V. 6. P. 362.
Xiu P., Xia Z., Zhou R. // Physical and Chemical Properties of Carbon Nanotubes. 2013. V. 8.
Butyrskaya E.V., Zapryagaev S.A., Izmailova E.A. Nechaeva L.S. // J. Phys. Chem. C. 2017. V. 121. P. 20524.
Frisch M. J., Trucks G.W., Schlegel H.B. et al. Gaussian 09. Gaussian, Inc., Wallingford CT, 2009.
Бутырская Е.В. Компьютерная химия: основы теории и работа с программами Gaussian и GaussView. М.: Солон–Пресс, 2011. 224 с.
Grimme S., Antony J., Ehrlich S. // J. Phys. Chem. 2010. V. 132. P. 154104.
Foresman J.B., Keith T.A., Wiberg K.B., Frisch M.J. // Ibid. 1996. V. 100. P. 16098.
https://www.vsu.ru/ru/news/feed/2017/03/8182.
Каплан И.Г. Введение в теорию межмолекулярных взаимодействий. М.: Наука, 1982. 311 с.
Шайтан К.В. Вычислительное и теоретическое моделирование биомолекулярных взаимодействий. Дубна, Россия, 2013. С. 62.
Ebadi A. // Adsorption. 2009. № 15. P. 65.
Раков Э.Г. Нанотрубки и фуллерены. М.: Университетская книга, 2006. 235 с.
Collins P.G. Defects and Disorder. NY.: Oxford University Press, 2010. P. 3193.
Нечаева Л.С., Бутырская Е.В., Запрягаев С.А. // Журн. cтруктур. химии . 2017. Т. 58. № 2. С. 233.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии