

Журнал физической химии, 2019, T. 93, № 6, стр. 856-865
Квантово-химическое моделирование влияния механоактивации растяжением на разрыв центральной С–С-связи в линейных полимерах
И. С. Флягина a, С. П. Долин a, b, А. И. Малкин a, В. И. Савенко a, *
a Российская академия наук, Институт физической химии и электрохимии им. А.Н. Фрумкина
Москва, Россия
b Российская академия наук, Институт общей и неорганической химии им. Н.С. Курнакова
Москва, Россия
* E-mail: visavenko@rambler.ru
Поступила в редакцию 10.01.2018
После доработки 04.12.2018
Принята к публикации 06.12.2018
Аннотация
Методами квантово-химического моделирования исследовано влияние механоактивации путем предварительного растяжения линейных полимерных углеводородных молекул (полиэтилена, полипропилена и др.) на энергетические и силовые характеристики процесса разрыва центральных С–С-связей в этих молекулах. Обнаружено, что в процессе деформирования С–С-связей происходит поэтапная перестройка электронной подсистемы молекул. Показано, что предварительная механоактивация полимерных молекул путем их однородного растяжения уменьшает энергию разрыва центральных С–С-связей в полимерном материале, что на качественном уровне согласуется с экспериментальными наблюдениями поведения полимерных макросистем подобного рода.
Механоактивация полимерных материалов – сложный, многоаспектный и иерархически организованный физико-химический процесс [1, 2]. Его результатом является изменение структурных характеристик материала, а, следовательно, и его механических, термодинамических и физико-химических свойств. В частности, помимо неизбежных структурных изменений, наблюдающихся в механоактивированном полимерном материале на макро- и мезоскопическом уровнях, в его молекулярных составляющих появляются значительные остаточные микро- и наноуровневые механические деформации и напряжения, существенно влияющие на прочностные характеристики продукта [3–7].
Экспериментальные исследования прочностных характеристик полимерных материалов требуют значительных экономических затрат и не всегда обеспечивают достаточно высокий прогностический уровень поведения изделий в реальных условиях их эксплуатации. Поэтому во многих конкретных случаях целесообразнее обратиться к теоретическому моделированию поведения соответствующих объектов. В связи с этим, в данной работе влияние предварительных (остаточных) растягивающих микродеформаций (микронапряжений) на энергосиловые аспекты процессов разрушения молекул линейных полимеров было изучено методами расчетной химии в рамках квантово-химического приближения.
МОЛЕКУЛЯРНЫЕ МОДЕЛИ МЕХАНОАКТИВИРОВАННЫХ ПОЛИМЕРОВ
Полимерный материал представлен простейшими элементарными звеньями различной длины – молекулами С6Н14 и С20Н42, первоначально находившимися в равновесном состоянии (рис. 1).
Рис. 1.
Статические равновесные структуры молекул C6H14 (а) и С20Н42 (б); обозначены начальные расстояния между концевыми атомами углерода (R0), между углеродными атомами центральной разрываемой связи (r), а также длины двух ближайших к ней С–С-связей (r1 и r2).
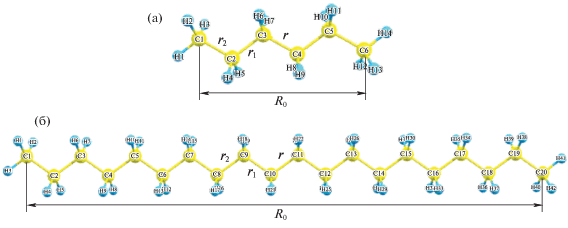
Остаточная микродеформация молекул ε, обусловленная предварительным механоактивационным растяжением полимера, задавалась посредством увеличения исходной длины R0 индивидуальной недеформированной молекулы с последующим закреплением ее концов на различных расстояниях (R): ε = ΔR/R0. Процесс разрыва углеродной цепи молекулы моделировали путем введения в предварительно микродеформированную систему центрального наноразмерного концентратора дополнительной микродеформации. Последняя задавалась путем последовательного увеличения расстояния r между соседними центральными углеродными атомами С–С в углеродной цепи при фиксированной постоянной остаточной микродеформации молекулы ε. Исследование влияния на поведение молекулярной системы именно центрального концентратора было обусловлено его наибольшей разрушительной способностью для полимера, поскольку несимметричные локальные концентраторы в молекулах реальных полимеров обычно характеризуются более высокими релаксационными свойствами [5].
В случае молекулы С6Н14 величина R варьировалась от его невозмущенного значения R0 = = 0.646 нм вплоть до деформированного Rε = = 0.696 нм с шагом 0.005 нм (что соответствует микродеформациям ε = ΔR/R0, лежащим в интервале значений ε от 0.77% до 7.7%), а в случае молекулы С20Н42 – от соответствующего равновесного расстояния R0 = 2.438 нм до значения Rε = = 2.498 нм с шагом 0.005 нм (при микродеформациях ε, лежавших в интервале значений 0.205–2.46%). Для получения равновесной геометрии микродеформированных молекул в каждом эксперименте была проведена полная энергетическая оптимизация всех системных параметров при различных фиксированных ε. В результате такой растягивающей микродеформации молекул все связи С–С в них равномерно удлинялись, а их направления благодаря увеличению валентных углов приближались к направлению продольной оси симметрии молекулы (скользящая ось симметрии второго порядка). Однако расчеты показали, что эти изменения составляли лишь малую долю от исходных величин вышеупомянутых характеристик благодаря весьма небольшим значениям ε, использованным в данной работе. Таким образом, предварительная микродеформация молекулярных структур не приводила к сколько-нибудь заметным качественным изменениям в их исходной конфигурации.
Для моделирования процесса разрыва центральной С–С-связи в обеих молекулах была использована процедура сканирования расстояния r от его равновесного значения (0.153 нм) при фиксированной величине шага (Δr = 0.03, 0.1 или 0.2 Å) до предельного значения r ≈ 0.75 нм, соответствующего полному разрыву деформируемой связи в молекуле. На каждом шаге сканирования также проводилась полная энергетическая оптимизация всех остальных геометрических параметров каждой системы (при фиксированных значениях параметров ε и r).
Энергии Ea, ε разрыва молекул были вычислены как разности между конечными значениями полной энергии системы при разрыве молекулы (Еdis) и значениями энергии этой же системы в точках энергетического минимума (Eeq, ε при Δr = = 0) на кривых Е = f(ε, r), описывавших изменение энергии системы в процессе удлинения центральной связи С–С: Ea, ε = Еdis – Еeq, ε.
Значения силы F(ε, r), действующей в молекулярных системах С6Н14 и С20Н42 при разрыве центральной связи С–С, вычислялись как отношение разности полной энергии системы ΔЕ(ε, r) = Е(ε, ri) – Е(ε, ri – 1) между двумя последовательными шагами ri и ri – 1 к соответствующему приращению межатомного расстояния Δr = (ri – ri – 1): F(ε, r) = ‒ΔЕ(ε, r)/Δr.
Дополнительно были рассчитаны распределения электронной плотности в заданных геометрических точках систем при различных значениях ε и r, а также теоретические колебательные спектры молекулы С6Н14 и продуктов ее разрушения. Эти данные по спектральным распределениям позволили получить количественные оценки изменений теплоемкости и энтропии в полимерной системе в результате деструкции составляющих ее молекул.
МЕТОДИКА ВЫЧИСЛЕНИЙ
Расчеты электронного и геометрического строения обеих молекул были выполнены с помощью вычислительного комплекса Gaussian 09 [8] и визуализированы с помощью программ GaussView 5.0 [9] и Chemcraft 1.7 [10]. Расчеты деформационного поведения энергосиловых характеристик обеих молекул были выполнены в рамках метода функционала плотности B3LYP, при этом в случае молекулы С6Н14 использовался базисный набор 6-311++G(d,p), а в случае молекулы С20Н42 – более компактный базис 6-31G(d,p). Вычисления колебательных частот в системе с получением соответствующих спектральных распределений, а также оценки величин эффективных зарядов на атомах также были выполнены с применением программы Gaussian 09 в рамках модели невозмущенных атомных орбиталей [11]. Численные значения электронной плотности в критических точках разрываемой центральной С–С-связи (определенных по методу Бейдера [12]) для молекулы С6Н14 были получены с помощью программы AIMALL [13].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Энергетические характеристики систем
Расчеты зависимостей полной энергии Е(ε, r) систем C6H14 и С20Н42 от расстояния r при различной остаточной деформации ε в системе показали следующее. В подавляющем большинстве случаев разрыв центральной С–С-связи происходит по гомолитическому механизму. При растяжении этой связи энергия системы увеличивается по закону, близкому к параболическому, стремясь в пределе к одной и той же величине для различных значений ε, а именно, к так называемому диссоционному пределу, с образованием слабосвязанного комплекса из двух фрагментов-радикалов С3Н$_{7}^{ \bullet }$ и С10Н$_{{21}}^{ \bullet }$ (см. рис. 2а, в). Однако при расчетах энергетического поведения системы C6H14 при ε = 3.9% и выше в некоторых случаях реализуется иной механизм расщепления указанных С–С‑связей, который можно условно назвать квазигетеролитическим. Соответствующие энергетические кривые демонстрируют резкие скачки в область малых (с учетом знака) энергий Е, вызванные диспропорционированием системы с образованием молекул C3H8 и С3Н6 (см. рис. 2в).
Рис. 2.
Зависимости полной энергии Е = f(ε, r) молекул C6H14 (а, б) и С20Н42 (в) от расстояния r при различных фиксированных значениях ε (а, в), (гомолитический разрыв), а также при значении ε = 3.9% для C6H14 (б), (квазигетеролитический разрыв). Во врезках показаны начальные участки энергетических зависимостей в увеличенном масштабе.

Более детализированные расчеты энергетического поведения системы с учетом возможности изменения в процессе разрыва молекул спиновой составляющей соответствующих волновых функций показали, что энергия Ea разрыва центральной связи в молекуле C6H14 с образованием двух одинаковых фрагментов-радикалов С3Н$_{7}^{ \bullet }$ равна Ea = 355 кДж/моль (ср. с данными [14]). Однако при образовании молекул C3H8 и С3Н6 она заметно уменьшается. Так, например, при ε = 3.9% она падает до величины Ea, ε = 3.9% = 314 кДж/моль (о степени достоверности этих цифр – см. ниже). Этот результат указывает на возможность перескока атома Н от одного фрагмента С3Н$_{7}^{ \bullet }$ к другому по “каналу” “квазиводородной” связи С‒Н···С (или электронного “мостика”), который реализуется при условии достаточного самопроизвольного (случайного) сближения любого углеродного (в том числе, центрального) и водородного атомов соседствующих фрагментов. Однако это возможно лишь при реализации в процессе разрыва молекулы большеугловых поворотов ее фрагментов относительно друг друга.
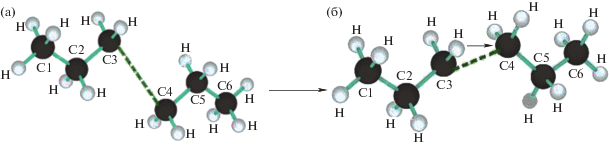
Расчеты показывают, что перескок атома Н происходит при межмолекулярном расстоянии С···С, равном 0.325 нм, которое оказывается “подходящим” для образования Н-связи такого типа с реализацией низкобарьерного двухъямного потенциала переноса протона. В качестве примера на рис. 3 приведен один из вариантов такого поворота. (Очевидно, что в молекулах, образующих полимерный материал, такие повороты возможны лишь в случаях разрушения на части микроблоков таких молекул как целого).
Рис. 3.
Структура молекулы С6Н14, до (а) и после (б) скачка на энергетической кривой Е = f(ε, r) при квазигетеролитическом разрыве молекулы (ε = 3.9%, Δr = 0.2 Å).
В отличие от молекулы С6Н14, для молекулы С20Н42 реализуется лишь гомолитический вариант разрыва С–С-связи и сколько-нибудь заметных разрывов на энергетических зависимостях Е = f(ε, r) не наблюдается (рис. 2б). Это связано с отсутствием возможностей для большеугловых случайных поворотов частей этой молекулярной системы при разрыве центральной С–С-связи благодаря значительной вращательной инерции ее гораздо более длинных углеродных полуцепей.
Учитывая найденную флуктуационно-статистическую неоднозначность формы потенциальных кривых, рассчитанных для молекулы С6Н14, с целью проверки устойчивости решений, выполненных с применением выбранной вычислительной схемы, а также достоверности полученных результатов, расчеты энергосилового поведения этой молекулы при ее разрыве были повторены для четырех различных значений ε = 0.00, 1.54, 2.32, 7.73% с измененным шагом варьирования расстояния r (∆r = 0.1 Å и ∆r = 0.2 Å). Результаты таких расчетов показали, что изломы на соответствующих энергетических кривых, несмотря на их случайный характер, наблюдаются тем чаще, чем больше величина шага ∆r, задаваемого расчетной схемой. Появление таких изломов, а также их величина ΔЕ, могут быть связаны как с алгоритмом поиска минимумов энергии используемой вычислительной программы, так и с величиной выбранного шага ∆r. При бόльшей величине шага геометрия молекулы С6Н14 при переходе между шагами изменяется сильнее, и поэтому найденные локальные энергетические минимумы системы также различаются сильнее. Возникающие при этом разрывы на энергетических кривых происходят, как правило, при таком изменении геометрии системы, которое включает повороты фрагментов молекулы вокруг разрываемой связи. При многочисленных повторных расчетах с наименьшим шагом ∆r = 0.1 отмеченное выше изменение взаимного расположения продуктов диссоциации происходило достаточно редко (не более 5% случаев), что указывает на малую вероятность его реализации.
(Альтернативное объяснение этим результатам может базироваться на учете особенностей метода DFT, алгоритм которого предопределяет сохранение на протяжении многих итераций качественно неизменного характера МО, построенных в схеме самосогласованного решения задачи [9, 11]. Можно предположить, что благодаря использованию такого алгоритма энергетически предпочтительное решение может появиться на более поздних стадиях расчета, а квазиравновесная энергия молекулярной системы может оказаться завышенной. Для подтверждения одной из указанных альтернативных возможностей в дальнейшем необходимо получить решение данной задачи с использованием иных неэмпирических методов, таких, например, как метод теории возмущений МР2).
Расчеты значений энергии разрыва центральной С–С-связи, проведенные в рамках использованного в данной работе подхода DFT в молекулах C6H14 и С20Н42 показали, что энергия разрыва этой связи, и, следовательно, полимерной системы как целого, во всех случаях понижается с ростом величины деформации ε по закону, близкому к квадратичному.
Из результатов этих расчетов также следует, что при остаточной деформации молекулы С6Н14, равной ε = 8% энергия Ea ее разрыва уменьшается на 8.1%, а при деформации молекулы С20Н42, равной ε = 2.4% энергия Ea уменьшается на 3.7%. Эти величины достаточно хорошо согласуются с экспериментальными данными, полученными при наблюдениях процессов разрушения реальных полимерных систем [3–7]. Вместе с тем, сопоставление соответствующих значений Ea, ε для рассмотренных двух молекулярных моделей показывает, что величины энергии разрыва центральных С–С-связей в них заметно различаются. Чтобы обнаружить возможное влияние длины молекулярной цепи на энергию разрыва ее центральной связи, дополнительно были проведены аналогичные серии расчетов для молекулярных моделей C2H6, C6H14, C10H22, C14H30, C16H34, C18H38, C30H62 и C40H82. Было найдено, что влияние остаточной деформации ε на энергию разрыва центральной С–С-связи в молекуле практически не зависит от длины ее углеродной цепи лишь для молекул, содержащих в цепи не менее десяти атомов углерода. Близкие по физическому смыслу результаты были получены ранее авторами работы [14].
Силовые характеристики систем
Более наглядное описание влияния степени растяжения центральной связи С–С на состояние полимерной системы можно получить из совместного анализа поведения функции Е(ε, r) и ее производной F(ε, r). Последняя имеет размерность силы, и в духе теоремы Фейнмана–Гельмана ее можно охарактеризовать как эффективную силу, включающую взаимодействие всех (а не только углеродных) атомов рассматриваемой цепочки. Вид этой функции для молекул C6H14 и C20H42 при некоторых значениях ε показан на рис. 4.
Рис. 4.
Сопоставление силовых зависимостей F(ε, r) для молекул С6Н14 (а) и С20H42 (б), рассчитанных для различных значений ε (см. легенду).

Эти зависимости качественно аналогичны полученным ранее авторами работы [15], в которой эффективные усредненные силы также были использованы для обсуждения механических свойств углеводородных молекул с различной длиной углеродной цепи.
Анализ данных, приведенных на рис. 3, показывает, что на любой силовой кривой F(ε, r) можно выделить две характерные области ее изменения как функции расстояния r. Первая из них описывает увеличение (по модулю) силы, возвращающей систему в равновесное состояние, а вторая – уменьшение этой силы с ее монотонным приближением к нулю, поскольку при r ≥ 0.5 нм связь С–С постепенно ослабляется, что соответствует гомолитическому расщеплению системы на два фрагмента С3Н$_{7}^{*}$ (в пределе – на соответствующие радикалы С3Н$_{7}^{ \bullet }$). Эти области разделяет критическая точка А*, в которой сила притяжения F(ε, r*) максимальна.
Согласно расчетам для обеих молекул критической точке А* соответствует критическое расстояние r* ≈ 0.2 нм, которое при изменении ε варьирует в довольно узком интервале (0.04 нм), оставаясь в любом случае близким к значению ван-дер-ваальсова радиуса атома углерода (0.18 нм) [16]. Это означает, что в первой области значений r превалируют валентные взаимодействия, а во второй – межмолекулярные. Из результатов расчетов также следует, что, несмотря на отмеченные выше флуктуации полной энергии Е = f(ε, r), связанные со случайными изменениями валентных и торсионных углов молекулы под влиянием наноконцентратора деформаций, а также с вариациями взаимной ориентации фрагментов при разрыве центральной связи, можно говорить о некоторой средней энергетической характеристике системы в критической точке, которая для обеих молекул в рамках рассматриваемого приближения составляет примерно 150 кДж/моль.
Рисунок 4 также иллюстрирует изменения в положении и глубине силовых минимумов, т.е. характеристик соответствующих силовых критических точек под влиянием остаточной деформации ε. По мере роста величины ε силовые минимумы смещаются в сторону бóльших расстояний r*, а их глубина показывает тенденцию к уменьшению. Объяснить это можно следующим образом. Несмотря на то, что каждом шаге расчетов величины ε и r фиксированы, геометрия остальной части цепи при минимизации энергии системы может релаксировать с заметными изменениями длин всех остальных, нефиксированных при расчете межъядерных расстояний, а также валентных и торсионных углов между нецентральными связями. Действительно, расчеты показывают, что в процессе разрыва молекул, с увеличением значений ε, растет не только первоначальная длина центральной С–С-связи. Заметно изменяются также длины двух С–С-связей, соседствующих с разрываемой. Хотя под действием наноконцентратора расстояние между центральными углеродными атомами монотонно увеличивается (при заданном постоянном значении общей остаточной деформации ε), длина прилегающих связей, локальные деформация εloc и напряжение в них при r < r* уменьшаются. Максимальная величина релаксации нецентральных связей С–С в системе, а также энергосиловое состояние всей системы, соответствующее критической точке на силовых кривых, с ростом величины ε достигается на более поздних стадиях деформирования центральной С–С-связи, т.е. при более значительных расстояниях r в системе. Это приводит к сдвигу значений r* в сторону их увеличения при росте остаточной микродеформации ε.
Уменьшение абсолютной величины силового минимума связано с тем, что полная энергия системы оказывается тем больше, чем больше остаточная деформация ε связей в молекуле (и напряжение в ней). Из данных, приведенных на рис. 4, также следует, что при переходе к гомологу с более длинной углеродной цепью эти эффекты ослабевают.
Изменение длин нецентральных С–С-связей в системе
Для обеих молекул были проанализированы полученные при расчетах релаксационные изменения длин связей r1 и r2 в зависимости от длины разрываемой связи r при различных значениях ε. Найдено, что при увеличении расстояния r в молекуле С6Н14 величины r1 и r2 сначала одновременно плавно уменьшаются вплоть до области критической точки А* на силовых кривых, а затем происходит скачкообразное удлинение связи r2. При дальнейшем увеличении расстояния r связь r1 укорачивается, а связь r2 продолжает удлиняться. При этом длины обеих связей стремятся к некоторому постоянному значению, равному значению длин этих связей в отдельных фрагментах после диссоциации исходной молекулярной системы. В молекуле С20Н42 имеет место аналогичный “дуализм” в поведении длин соседних связей, однако в этом случае в системе почти не наблюдаются энергетические и силовые скачкообразные флуктуации. Эти результаты подтверждают справедливость постулата о ключевой роли релаксации межатомных связей в энергосиловом поведении молекул полимеров в процессе их разрушения [5].
Изменения геометрии углеродной цепочки молекул С6Н14 и С20Н42 в процессе разрыва центральной С–С-связи при различных остаточных деформациях ε углеродной цепи можно объяснить в рамках вибронной модели гетеролигандных систем (ГС) [17, 18]. В вибронной модели ГС при анализе энергетического состояния системы учитываются электронно-колебательные эффекты, обусловленные смешиванием электронных состояний благодаря ядерным смещениям (электрон-фононное взаимодействие). Это приводит к появлению в соответствующих спектрах неполносимметричных нормальных колебаний, заметно влияющих на квазиравновесную геометрию системы и ее энергосиловые характеристики.
В рамках этой модели структуру углеводородного олигомера можно рассматривать как совокупность гетеролигандных (квази)тетраэдрических систем состава СН2ХХ', где Х, Х' = СН3- или СН2-лиганды. В вибронной модели таких систем смещения атомов Н в молекуле СН4 эквивалентны, и замещение атомов Н группами с другой электроотрицательностью позволяет записать в аналитическом виде обобщенные силы, приводящие к смещению всех атомов относительно их исходных равновесных положений [17, 18], а, следовательно, найти взаимные атомные смещения в системе. Анализ структурных характеристик молекул, проведенный для ряда систем с различной длиной цепи (CH4, C3H8, C(CH3)4) в рамках квантово-химических приближений разного уровня (B3LYP, MP2, QCISD) полностью подтверждают применимость вибронной модели к описанию структурных изменений в углеводородных олигомерах.
Рассчитанные в рамках вибронной модели [17, 18] смещения атомов углеродной цепи в рассматриваемых нами молекулярных системах позволили найти знаки и величины элементарных сил, действующих на любой атом цепочки при ее деформировании и разрыве (см. рис. 5).

Электронная плотность
При моделировании влияния остаточной микродеформации растяжения ε на разрыв молекул С6Н14 и С20Н42 представляет интерес проанализировать сопутствующие этому процессу изменения в распределении электронной плотности в данных молекулах и установить взаимосвязь этих изменений с вариациями геометрических характеристик углеродной цепи. Для этой цели расчеты электронной плотности были выполнены, прежде всего, в наиболее характерной точке системы – в точке симметрии разрываемой центральной С–С-связи. Естественно, что для рассматриваемых случаев эта точка совпадает с серединой расстояния между соединенными этой связью центральными атомами углерода. Изменения электронной плотности в этой “симметрийной” точке в зависимости от относительного удлинения ε и расстояния r, рассчитанные для молекулы С6Н14, приведены на рис. 6. Видно, что электронная плотность в “симметрийной” точке линейно уменьшается с ростом остаточной микродеформации ε молекулы. Расчеты также показывают, что указанная электронная плотность почти линейно уменьшается при увеличении расстояния r вплоть до полного разрыва связи, при котором она приближается к нулю.
Рис. 6.
Электронная плотность в “симметрийной” точке разрываемой С–С-связи молекулы С6Н14 как функция от: (а) микродеформации молекулы ε в равновесном состоянии до начала процесса разрыва (при ∆r = 0) и (б) от расстояния r при различных величинах микродеформации ε (см. легенду).

Следует пояснить, что на рис. 6б, представлены результаты расчетов электронной плотности в симметрийной точке не для всех возможных значений расстояния r, а лишь для тех его значений, которые соответствуют избранным характерным энергетическим состояниям молекулы С6Н14, а именно: для равновесного состояния (первый шаг расчетов), для силовой критической точки А*(r*), соответствующей минимуму на силовых кривых: F(ε, r*), для значений параметра r, соответствовавших диссоционному пределу (образование слабо связанных одинаковых фрагментов), а также для тех значений r, при которых фиксировались значительные флуктуации на силовых кривых. Наблюдающийся во всех изображенных на рис. 6б случаях одинаковый квазилинейный характер уменьшения значений электронной плотности с ростом величины r указывает на то обстоятельство, что распределение электронной плотности в пространстве между разрываемыми атомами зависит только от длины разрываемой связи (точнее, от ее локальной деформации kΔr/r0, где k – число шагов в процессе ее локального удлинения) и не зависит от других изменений в геометрии системы, т.е. от значений валентных и двугранных углов в молекуле и величины ее общей остаточной деформации ε.
Вычисления зарядов на атомах углеродной цепи методом невозмущенных орбиталей (ЛКАО) [11, 13] показали, что в процессе разрыва центральной С–С-связи отрицательный заряд, и, соответственно, эффективная электронная плотность, локализованная на центральных атомах углерода, соединенных разрываемой связью, уменьшается, а на двух ближайших к ним соседних углеродных атомах возрастает (см. рис. 7).
Рис. 7.
Зависимости эффективной электронной плотности на центральных углеродных атомах, а также на атомах, соседствующих с ними, от длины центральной связи r.

Последнее означает, что при разрыве молекул С6Н14 и С20Н42 часть заряда переходит от разрываемой связи на две соседние; при этом изменения в распределении заряда на двух последующих связях оказываются относительно небольшими. Вместе с тем, расчеты для системы С20Н42, в которой углеродная цепочка существенно длиннее, чем в молекуле С6Н14, показали, что распределение зарядов на атомах углерода, достаточно удаленных от разрываемых атомов, практически не изменяются.
Таким образом, хотя обусловленное разрывом центральной С–С-связи возмущение системы приводит к альтернированию длин связей углеродной цепочки, но оно затрагивает только две ближайших связи в каждом фрагменте. Этот вывод полностью согласуется с наблюдаемыми изменениями длин связей в цепочке и находит адекватное обоснование в рамках вышеуказанной вибронной модели [17, 18].
Колебательные спектры
Колебательные спектры для молекулы С6Н14 были получены в точках равновесия, силовых критических точках r* и точках “диссоционного” предела для всех значений остаточной деформации ε, а также для фрагмента С3Н7 и ионов С3Н$_{7}^{ + }$ и С3Н$_{7}^{ - }$. Как известно [19], колебательные спектры фрагментов С3Н7 и С6Н14 имеют 24 и 54 частоты соответственно, 7 и 14 из которых связаны с колебаниями валентных связей С–Н. Частоты (волновые числа λ) колебаний С–Н-связей в молекуле С6Н14 имеют высокие значения, локализованные вблизи λ = 3000 см–1 и при микродеформировании системы, как и следовало ожидать, практически не меняются. Это отличает их поведение от поведения частот колебаний С–С-связей и может служить некоторым обоснованием приемлемости использованной методики анализа частотных характеристик системы. Рассчитанные в рамках нормально-координатного приближения спектральные интенсивности колебаний аниона С3Н$_{7}^{ - }$ в среднем превышают соответствующие колебательные интенсивности нейтрального фрагмента С3Н7 и катиона С3Н$_{7}^{ + }$ более чем в 10 раз, что указывает на гораздо более высокие значения колебательной теплоемкости и колебательной энтропии радикала С3Н$_{7}^{ + }$ по сравнению с двумя другими продуктами диссоциации молекулы С6Н14. Однако этот неочевидный вывод нуждается в прямом экспериментальном подтверждении.
Наибольший интерес представляют колебания разрываемой C–C-связи, которым соответствуют несколько частот расчетного колебательного спектра молекулы С6Н14. Частоты этих колебаний уменьшаются с увеличением микродеформации растяжения ε и, соответственно, остаточных растягивающих напряжений в данной молекулярной системе. Этот вывод находится в согласии с качественной оценкой изменения силовых констант данной связи при ее растяжении. Полученные результаты позволяют приближенно оценить изменения колебательной теплоемкости и энтропии при однородной микродеформации и диссоциации молекулы С6Н14. Для колебательной теплоемкости Cvib и энтропии Svib при достаточно низких гомологических температурах T, когда изменения в частотном спектре сказываются на термодинамичских характеристиках системы наиболее ощутимо, справедливы соотношения [20]:
ЗАКЛЮЧЕНИЕ
Установлено, что энергия разрыва межатомных С–С-связей в молекулярных олигомерах, моделирующих компоненты линейных полимерных материалов, уменьшается с увеличением вызванной механоактивацией остаточной микродеформации (микронапряжения) в системе. При относительном удлинении олигомеров С6Н14 и С20Н42 на 8% и 2.4% расчетная энергия их разрыва уменьшается, соответственно, на 8.1 и 3.7%. Иными словами, механоактивация может ощутимо понизить разрывную прочность соответствующего линейного полимерного материала.
Показано, что в процессе разрыва межатомных связей в углеводородных олигомерах происходит поэтапная перестройка их электронной подсистемы. На первом этапе, при малых микродеформациях разрываемых межатомных связей, в электронной подсистеме молекул возникают процессы перераспределения электронной плотности, препятствующие ее разрушению (правило Ле-Шателье–Брауна). На втором этапе, при относительно больших локальных деформациях разрываемых связей, преобладают изменения в электронной конфигурации молекулярной системы, ведущие к ее деструкции.
Теоретически рассчитанные колебательные спектры олигомерных молекулярных систем, моделирующих полимеры, показывают уменьшение частот колебаний разрываемой связи по мере увеличения остаточной микродеформации системы в результате ее механоактивации. Благодаря этому существенно меняются также и термодинамические характеристики соответствующих молекулярных систем. Можно ожидать, что в результате механоактивации полимерного материала как целого таким же образом будут меняться его термодинамические свойства.
Результаты выполненных расчетов качественно согласуются с многочисленными экспериментальными наблюдениями влияния механоактивации на физико-химические, механические и термодинамические характеристики полимерных материалов [1, 3–7].
Список литературы
Аввакумов Е.Г. Фундаментальные основы механической активации, механосинтеза и механохимических технологий. Новосибирск: Наука, 2009. 344 с.
Панин В.Е., Лихачев В.А., Гриняев Ю.В. Структурные уровни деформации твердых тел. Новосибирск: Наука, 1985. 255 с.
Аскадский А.А. Деформация полимеров. М.: Химия, 1973. 448 с.
Гуль В.Е. Структура и прочность полимеров. М.: Химия, 1978. 328 с.
Бартенев Г.М. Прочность и механизм разрушения полимеров. М.: Химия, 1984. 280 с.
Нарисава И. Прочность полимерных материалов. М.: Химия, 1987. 400 с.
Дубинская А.М. // Успехи химии. 1999. Т. 68. № 8. С. 708.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09 // Gaussian Inc. Wallingford. CT. USA: 2009. revision D.01.
Dennington R., Keith T. Millam. GaussView. Version 5. // J. Semichem Inc. Shawnee Mission Ks. 2009.
Zhurko G.A., Zhurko D.A. Chemcraft. // www.chemcraftprog.com. 2015.
Glendening E.D., Reed A.E., Carpenter J.E., Weinhold F. NBO 6.0 Program Manual. // NBO6 Website: http://nbo6.chem.wisc.edu. 2013.
Бейдер Р. Атомы в молекулах. Квантовая теория. М.: Мир, 2001. 532 с.
Keith T.A. AIMALL. http://aim.tkgristmill.com. 2015.
Alkorta I., Elguero J. // Chem. Phys. Lett. 2006. V. 425. № 4–6. P. 221.
Yushchenko V.S., Ponomareva T.P., Shchukin E.D. // J. Mater Sci. 1992. V. 27. № 6. P. 1659.
Бацанов С.С. // Неорган. материалы. 2001. Т. 37. № 9. С. 1031.
Левин А.А., Долин С.П. // Координац. химия. 1979. Т. 5. № 3. С. 320.
Левин А.А., Дьячков П.Н. Электронное строение, структура и превращения гетеролигандных молекул. М.: Наука, 1990. 256 с.
Encyclopedia of Spectroscopy and Spectrometry. Lindon J.-2nd Ed. Academic Press, 2010. 3312 p.
Ландау Л.Д., Лифшиц Е.М. Статистическая физика. М.: Физматлит. 2005. 616 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии