

Журнал физической химии, 2019, T. 93, № 6, стр. 913-923
Влияние концентрации органического модификатора водно-этанольной подвижной фазы на удерживание и термодинамику адсорбции энантиомеров гидроксикислот и их производных на хиральном адсорбенте с привитым антибиотиком эремомицином
О. Ш. Гоголишвили a, *, Е. Н. Решетова a, **
a Российская академия наук, Уральское отделение, Институт технической химии
614013 Пермь, Россия
* E-mail: otar5993@mail.ru
** E-mail: lenire@yandex.ru
Поступила в редакцию 27.08.2018
После доработки 10.10.2018
Принята к публикации 17.10.2018
Аннотация
Исследованы закономерности удерживания и термодинамика адсорбции энантиомеров ароматических гидроксикислот и их производных на хиральной неподвижной фазе с привитым макроциклическим антибиотиком эремомицином из водно-этанольных элюентов в условиях линейной жидкостной хроматографии. Выявлены зависимости характеристик удерживания, разделения и термодинамических величин от концентрации органического модификатора в элюенте. Показана взаимосвязь строения исследуемых адсорбатов с удерживанием и селективностью разделения их энантиомеров. Продемонстрировано, как экстратермодинамические корреляции могут быть использованы для получения информации о механизме адсорбции. Установлено различие механизма адсорбции энантиомеров гидроксикислот и их эфиров, а также гидроксикислот, имеющих противоположный порядок элюирования S- и R-энантиомеров.
Гидроксикислоты (ГК) представляют собой один из наиболее важных классов хиральных соединений в фармацевтической, косметической [1] и агрохимической промышленностях [2]. Подавляющее большинство ГК и их производных проявляют биологическую активность и требуются в виде индивидуальных оптических изомеров. Энантиомерно чистые α-ГК применяются в качестве прекурсоров для производства хиральных лекарственных соединений [3, 4], биоразлагаемых полимеров [5], сольватирующих и разделяющих агентов [6]. R-Миндальная кислота является основным промежуточным соединением для получения полусинтетических пенициллинов и цефалоспоринов, используется для синтеза противоопухолевых, снижающих вес препаратов и других фармацевтических соединений [7, 8]. S-Миндальная кислота может применяться для получения стабильной сокристаллической твердой формы антиангинального препарата гидрохлорида ивабрадина [9].
Эффективным методом разделения энантиомеров является высокоэффективная жидкостная хроматография на хиральных неподвижных фазах (ХНФ). Одними из наиболее перспективных ХНФ, используемых в жидкостной хроматографии для разделения энантиомеров биологически активных соединений, являются хиральные адсорбенты на основе привитых к поверхности силикагеля макроциклических гликопептидных антибиотиков [10]. Молекулы антибиотиков содержат множество функциональных групп и хиральных центров (ХЦ), обеспечивающих успешное разделение энантиомеров различных классов соединений, в особенности, содержащих карбоксильные группы [11–14]. Одним из таких адсорбентов является разработанная российскими учеными [15] ХНФ “Nautilus-E” с привитым к поверхности силикагеля макроциклическим антибиотиком эремомицином. Указанная ХНФ продемонстрировала разделяющую способность по отношению к энантиомерам аминокислот [12, 15, 16], профенов [17, 18] и α-фенилкарбоновых кислот [19, 20]. Общей чертой изученных соединений является наличие карбоксильной группы и ароматического кольца. Карбоксильная группа создает возможность образования дипольных взаимодействий и водородных связей, а ароматическая часть молекул способствует образованию π–π-комплексов между адсорбатом и неподвижной фазой (НФ). Наличие обеих групп в молекуле адсорбата позволяет реализовывать наиболее сильное взаимодействие адсорбата с эремомицином [16]. При этом энантиораспознавание усиливается, если разделяемое соединение содержит карбонильную группу или ароматическое кольцо в непосредственной близости к стереоцентру (т.е. в α-, β-, или γ-положении) [21].
Проявление хиральным адсорбентом “Nautilus-E” высокой селективности разделения по отношению к энантиомерам миндальной и α-метоксифенилуксусной кислот [19, 20] обусловило интерес к подробному изучению хроматографического поведения энантиомеров ГК и их производных с различным набором заместителей у ХЦ. Закономерности удерживания и механизм энантиораспознавания в этом случае могут различаться для ГК и их производных. Выявить особенности механизмов удерживания и энантиораспознавания позволяет анализ термодинамических зависимостей при адсорбции энантиомеров родственных соединений.
Целью настоящего исследования являлось изучение влияния концентрации органического модификатора в элюенте на характеристики удерживания, разделения и термодинамику адсорбции энантиомеров ГК и их производных на ХНФ с привитым антибиотиком эремомицином. В качестве тестовых адсорбатов выбраны энантиомеры ГК и их замещенных: миндальной (1), 2‑хлорминдальной (2), 2-гидрокси-2-фенилпропановой (атромолочной) (3), 2-гидрокси-3-фенилпропановой (4), α-метоксифенилуксусной (5), 3-гидрокси-3-фенилпропановой (9), α-ацетилминдальной (10) и α-метокси-α-(трифторметил)фенилуксусной (кислота Мошера) (11), а также энантиомеры эфиров миндальной кислоты: метилового (6), этилового (7) и бензилового (8) (табл. 1)
Таблица 1.
Характеристики удерживания (k'), селективности разделения (α) и разрешения пиков (RS) энантиомеров гидроксикислот и их производных в элюенте с концентрацией этанола 80 об. %, T = 22°C
| № | Соединение | Формула | k′ | α | RS | |
|---|---|---|---|---|---|---|
| S | R | |||||
| 1 | Миндальная кислота |  |
3.41 | 4.81 | 1.41 | 4.08 |
| 2 | 2-Хлорминдальная кислота |  |
3.65 | 5.52 | 1.51 | 7.70 |
| 3 | 2-Гидрокси-2-фенил-пропановая кислота |  |
3.06 | 3.32 | 1.09 | 0.96 |
| 4 | 2-Гидрокси-3-фенил-пропановая кислота |  |
2.60 | 9.35 | 3.60 | 15.87 |
| 5 | α-Метоксифенилуксусная кислота | 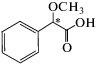 |
2.58 | 6.34 | 2.46 | 17.71 |
| 6 | Метиловый эфир миндальной кислоты |  |
0.13 | 0.13 | 1.03 | 0.13 |
| 7 | Этиловый эфир миндальной кислоты |  |
0.09 | 0.10 | 1.03 | 0.11 |
| 8 | Бензиловый эфир миндальной кислоты | 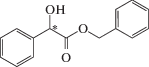 |
0.08 | 0.10 | 1.15 | 0.30 |
| 9 | 3-Гидрокси-3-фенил-пропановая кислота |  |
1.86 | 1.42 | 1.31 | 5.14 |
| 10 | α-Ацетилминдальная кислота |  |
2.36 | 2.20 | 1.07 | 1.42 |
| 11 | α-Метокси-α-трифторметилфенилуксусная кислота |  |
1.17 | 1.16 | 1.01 | 0.14 |
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Оборудование. Эксперименты выполнены на жидкостном хроматографе Agilent 1100 Series (Agilent Technologies, США), оснащенным прецизионным насосом, диодно-матричным детектором, автоматическим дозатором и термостатом колонок, обеспечивающим постоянство температуры (±0.2 К). Для дегазации подвижных фаз (ПФ) использовалась ультразвуковая ванна УЗВ–3/100–ТН–РЭЛТЕК.
Хиральная неподвижная фаза. Хроматографическая колонка (250 × 4.6 мм) производства ЗАО “БиоХимМак СТ” заполнена силикагелем с привитым макроциклическим гликопептидным антибиотиком эремомицином. Диаметр частиц сорбента 5 мкм.
Реагенты. Для приготовления элюентов использовали уксусную кислоту и ацетат аммония квалификации “х.ч.”, бидистиллированную воду и ректификованный этанол (96%, “высш. оч.”).
В исследовании использовались энантиомеры миндальной, α-метоксифенилуксусной, 2-гидрокси-3-фенилпропионовой, 3-гидрокси-3-фенилпропионовой кислот, R-энантиомеры 2-хлорминдальной, α-метокси-α-(трифторметил)фенилуксусной, 2-ацетилминдальной кислот, S‑энантиомер 2-гидрокси-2-фенилпропионовой кислоты фирмы “Alfa Aesar”, S-энантиомер 2‑хлорминдальной кислоты, R-энантиомер 2‑гидрокси-2-фенилпропионовой кислоты, энантиомеры метилового, этилового и бензилового эфиров миндальной кислоты фирмы “Sigma–Aldrich”.
Методика работы. В качестве ПФ использовали смеси водного 0.1 М ацетата аммония (с добавкой уксусной кислоты 0.175 М) с этанолом в соотношениях 90 : 10, 60 : 40, 40 : 60, 30 : 70, 20 : 80, 10 : 90 ($v{\text{/}}v$). Влияние концентрации этанола в элюенте на термодинамические характеристики адсорбции исследовали с ПФ составов ацетатный буфер–этанол 60 : 40, 40 : 60, 20 : 80.
Температурный диапазон исследований 22–50°С. Скорость потока элюента 0.5 мл/мин. Концентрация исследуемых аналитов 0.5 мг/мл. Объем вводимой пробы 2 мкл. Детектирование энантиомеров всех исследуемых соединений осуществляли при длине волны 254 нм. Каждое измерение повторяли 3 раза. Для определения мертвого времени использовали три-трет-бутилбензол [22].
Термодинамические характеристики адсорбции (стандартную энтальпию ΔH0 и энтропию ΔS0) определяли по уравнению Вант-Гоффа, пользуясь линейной зависимостью между логарифмом фактора удерживания k' и обратной температурой. Для регистрации хроматограмм и обработки результатов использовалась программа ChemStation Rev.A.08.03Rus.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Влияние структуры адсорбатов на удерживание и энантиоселективность разделения. Исследуемые соединения – ароматические α- и β-ГК и их производные отличаются строением заместителей у ХЦ (табл. 1), поэтому закономерности удерживания и селективности разделения их оптических изомеров будут определяться природой всех заместителей в функциональных группах и при ХЦ молекул ГК и их производных.
Сравнение хроматографических характеристик энантиомеров соединений 1–11 (табл. 1) показало, что S-энантиомер адсорбатов 1–8 элюируется первым. Для энантиомеров кислот 9–11 наблюдается изменение порядка элюирования (табл. 1). Для кислоты 9, очевидно, это связано с β-положением гидрокси-группы к карбоксильной группе. На инверсию порядка элюирования энантиомеров кислот 10 и 11, вероятно, влияют стерические затруднения, создаваемые сложноэфирной и –CF3-группами в соответствующих структурах молекул.
По фактору удерживания (k') первого элюируемого S-энантиомера адсорбаты 1–8 располагаются в ряду: 8 < 7 < 6 < 5 < 4 < 3 < 1 < 2 (табл. 1). Наиболее сильное удерживание S-энантиомеров кислот 1–4 обусловлено наличием в α-положении у ХЦ молекул полярных гидрокси- и карбокси-групп, способных к взаимодействию с активными центрами хирального селектора (ХС). Этерификация карбоксильной группы приводит к снижению k' энантиомеров эфиров (8 < 7 < 6 < 1) и энантиоселективности разделения (α) 6 – 8, вплоть до полной ее потери для 6 и 7 (табл. 1), что свидетельствует о значительном вкладе в механизм энантиораспознавания ионообменных взаимодействий между карбокси-группой адсорбата и полярными группами ХС [14]. Более высокие значения α для эфира 8, очевидно, связаны с дополнительным вкладом π–π-взаимодействий бензильного заместителя с ароматическими кольцами ХС.
Метилирование гидроксильной группы у ХЦ приводит к более слабому удерживанию S-энантиомеров: 5 < 1, но способствует росту α для кислоты 5 по сравнению с кислотой 1 (табл. 1), в то же время ее ацилирование ведет к потере селективности разделения кислоты 10 (табл. 1). Данный феномен можно объяснить изменением электронодонорной способности заместителя [23] и потенциального участка водородной связи [6], что способствует хиральному распознаванию, либо ограничивает его.
При 80 об. % этанола в элюенте удлинение боковой цепи в ряду S-α-ГК (4 > 1) на группу –CH2 (табл. 1) приводит к уменьшению факторов удерживания k' (1) > k' (4), что обусловлено снижением прочности ионных взаимодействий между ХС и адсорбатом при усилении его гидрофобноcти, поскольку при высоких концентрациях органического модификатора в элюенте удерживание на ХНФ “Nautilus-E” определяется, преимущественно, сильными ионными взаимодействиями [13]. При уменьшении концентрации этанола в ПФ до 40 об. % (и ниже) удерживание ГК описывается закономерностями обращенно-фазовой (ОФ) ВЭЖХ и определяется, главным образом, гидрофобными взаимодействиями между адсорбатом и макроциклическим антибиотиком, поэтому увеличение длины боковой цепи на группу –CH2, напротив, ведет к более сильному удерживанию S-энантиомеров : k' (1) < k' (4) (рис. 1а).
Рис. 1.
Зависимости фактора удерживания k' (а), селективности разделения α и разрешения пиков RS (б) энантиомеров гидроксикислот и их производных от концентрации этанола в подвижной фазе при 22°С.
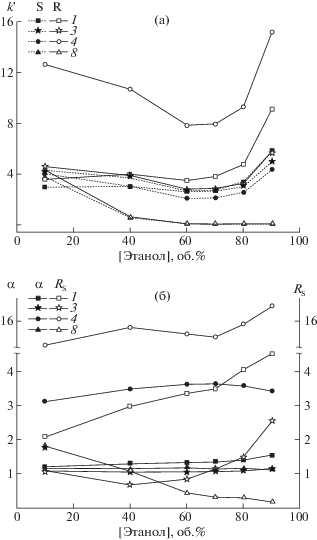
Представленная последовательность элюирования S-энантиомеров нарушается для сильнее удерживаемых R-энантиомеров адсорбатов: 8 ≈ ≈ 7 < 6 < 3 < 1 < 2 < 5 < 4 (табл. 1), что может свидетельствовать о различном механизме удерживания S- и R-оптических изомеров. Наиболее сильное удерживание наблюдается для R-энантиомера кислоты 4 (табл. 1). Для нее разность $k_{{\text{R}}}^{'} - k_{{\text{S}}}^{'}$, отражающая отличия в удерживании энантиомеров за счет селективного связывания составляет 72% от величины $k_{{\text{R}}}^{'}$ и слабо зависит от условий эксперимента. Очевидно, строение центра хирального распознавания селектора предпочтительно для образования адсорбционного комплекса с R-конфигурацией α-ГК, содержащих –CH2-группу между ароматическим кольцом и ХЦ, поскольку R-энантиомер кислоты 1, гомологичного строения, удерживается слабее и энантиоразделение характеризуется невысокими значениями α (табл. 1). Аналогичные результаты были получены авторами [12] для адсорбции энантиомеров аминокислот на ХНФ “Nautilus-E”. Поэтому логично допустить, что центром хирального распознавания для ГК, как и для аминокислот, является гидрофобный “карман”, локализованный между углеводными остатками эремомицина [12].
Наличие у ХЦ заместителей, создающих стерические затруднения (кислоты 3 и 11) приводит к полной потере энантиоселективности разделения (табл. 1). Кроме того, длина связи С*–CH3 и С*–CF3 у ХЦ молекул кислот 3 и 11 равна 1.5 Å и превышает длину симметрично расположенной связи C*–Н (1.1 Å) у ХЦ молекул остальных исследуемых адсорбатов. Это обстоятельство может являться причиной потери энантиоразделения этих кислот.
Влияние концентрации органического модификатора в элюенте на удерживание и энантиоселективность. На рис. 1а приведены зависимости фактора удерживания энантиомеров кислот 1, 3, 4 и эфира 8 от концентрации этанола в элюенте. Зависимость k' энантиомеров ГК 1, 3 и 4 от содержания этанола в элюенте имеет экстремальный характер с минимумом при 60 об. % этанола (рис. 1а). Это связано с тем, что при содержании органического модификатора в элюенте до 60 об. % реализуется истинно ОФ вариант хроматографии: с понижением полярности элюента повышается его способность к вытеснению молекул адсорбатов с активных центров НФ, что ведет к снижению k' энантиомеров кислот. Дальнейший рост концентрации органического модификатора в элюенте приводит к заполнению им полостей ХС эремомицина [13]. Удерживание энантиомеров ГК в этом случае определяется более сильными ионными взаимодействиями, на что указывает восходящая ветвь зависимости (рис. 1а). Подобный характер зависимости наблюдался для удерживания энантиомеров аминокислот [13, 24, 25] и ароматических аминов [26] на ХНФ с привитыми антибиотиками тейкопланином [24, 25], ванкомицином [26] и эремомицином [13].
Состав ПФ почти не влияет на энантиоселективность разделения ГК 1, 3, 4 (рис. 1б). Фактор разрешения пиков (RS) более чувствителен к изменению состава элюента. При увеличении содержания этанола в элюенте от 10 до 90 об. % RS энантиомеров кислот 1, 3 и 4 заметно увеличивается и наблюдается слабая тенденция к увеличению α кислот 1 и 3, что указывает на лучшую растворимость кислот в этаноле. Вероятно, электростатические силы, отвечающие за хиральное распознавание ГК, ослабевают в обогащенных водой фазах [24]. Аналогичные закономерности были получены для аминокислот и их производных на ХНФ с тейкопланином [14, 24]. В работе [12], напротив, обнаружено снижение α аминокислот на ХНФ “Nautilus-E” при увеличении концентрации органического модификатора в элюенте. Авторы [12] объяснили это реализацией взаимодействий сорбатов с неселективными центрами в антибиотике.
Для эфира 8 зависимости факторов k' (рис. 1а), α и RS от концентрации этанола в элюенте (рис. 1б) имеют монотонно-убывающий характер, указывая на ослабевание ионных взаимодействий с полярными группами селектора.
Влияние концентрации этанола в элюенте на термодинамические характеристики адсорбции. На основании линейного характера температурных зависимостей фактора удерживания энантиомеров всех исследуемых соединений (коэффициенты корреляции r2 > 0.97) определены термодинамические характеристики адсорбции по уравнению Вант-Гоффа, которое для величины k' записывается следующим образом:
(1)
$\ln k{\kern 1pt} ' = - \frac{{\Delta H_{{}}^{0}}}{{RT}} + \frac{{\Delta S_{{}}^{0}}}{R} + \ln \varphi ,$Параметр φ, как правило, сложно определить с высокой точностью, поскольку его значение может изменяться с температурой [22, 27]. Однако линейность температурной зависимости факторов удерживания свидетельствует о стабильности механизма удерживания, отсутствии структурных трансформаций ХС в исследованном диапазоне температур [11] и инвариантности значений ΔH0 и ΔS0 при изменении температуры [28].
В табл. 2 приведены значения термодинамических величин адсорбции энантиомеров ГК и их производных для разных составов ПФ. Наблюдается общая тенденция к снижению теплового эффекта адсорбции S- и R-энантиомеров соединений 1–11 с увеличением доли этанола в ПФ от 40 до 80 об. %. Однако характер зависимостей термодинамических величин ΔH0 и ΔS0 от концентрации этанола в элюенте различен для энантиомеров кислот (1–5, 9–11) и эфиров (6–8), что может свидетельствовать о различии механизма адсорбции ионогенных и нейтральных соединений на исследуемой ХНФ (табл. 2).
Таблица 2.
Термодинамические характеристики адсорбции (кДж/моль) энантиомеров гидроксикислот и их производных при различной концентрации этанола в подвижной фазе
| [Этанол], об. % | –ΔH0 | –TΔS0 | ΔG0 | Δ(ΔH0) | TΔ(ΔS0) | –Δ(ΔG0) | Tiso, град | |||
|---|---|---|---|---|---|---|---|---|---|---|
| S | R | S | R | S | R | |||||
| 1 | ||||||||||
| 40 | 14.07 | 15.70 | 10.73 | 11.77 | –3.34 | –3.93 | –1.63 | –1.04 | 0.59 | 211 |
| 60 | 11.29 | 13.09 | 8.23 | 9.37 | –3.06 | –3.72 | –1.80 | –1.14 | 0.66 | 213 |
| 80 | 9.37 | 11.60 | 5.55 | 7.00 | –3.82 | –4.60 | –2.23 | –1.45 | 0.78 | 200 |
| 2 | ||||||||||
| 40 | 14.57 | 16.10 | 10.60 | 11.76 | –3.97 | –4.34 | –1.53 | –1.16 | 0.37 | 133 |
| 60 | 11.40 | 13.71 | 8.01 | 9.80 | –3.39 | –3.92 | –2.31 | –1.79 | 0.52 | 126 |
| 80 | 9.35 | 14.12 | 5.36 | 9.34 | –3.99 | –4.79 | –4.77 | –3.98 | 0.79 | 97 |
| 3 | ||||||||||
| 40 | 15.81 | 16.18 | 12.02 | 12.32 | –3.78 | –3.86 | –0.37 | –0.30 | 0.07 | 111 |
| 60 | 12.17 | 12.47 | 9.12 | 9.30 | –3.06 | –3.18 | –0.30 | –0.18 | 0.12 | 243 |
| 80 | 9.70 | 10.05 | 6.18 | 6.33 | –3.52 | –3.72 | –0.35 | –0.15 | 0.20 | 442 |
| 4 | ||||||||||
| 40 | 15.44 | 28.31 | 12.19 | 22.44 | –3.25 | –5.87 | –12.87 | –10.25 | 2.62 | 114 |
| 60 | 11.14 | 25.17 | 8.62 | 19.98 | –2.52 | –5.19 | –14.03 | –11.36 | 2.67 | 108 |
| 80 | 8.70 | 23.99 | 5.55 | 18.27 | –3.15 | –5.72 | –15.29 | –12.72 | 2.57 | 98 |
| 5 | ||||||||||
| 40 | 11.09 | 18.00 | 8.57 | 13.60 | –2.52 | –4.39 | –6.91 | –5.03 | 1.87 | 150 |
| 60 | 8.38 | 16.04 | 6.14 | 11.83 | –2.24 | –4.20 | –7.66 | –5.70 | 1.96 | 142 |
| 80 | 6.50 | 14.34 | 3.26 | 9.16 | –3.24 | –5.18 | –7.85 | –5.90 | 1.94 | 137 |
| 6 | ||||||||||
| 40 | 13.48 | 13.63 | 15.90 | 16.01 | 2.42 | 2.38 | –0.15 | –0.11 | 0.04 | 160 |
| 60 | 12.20 | 12.11 | 16.65 | 16.50 | 4.45 | 4.39 | 0.09 | 0.15 | 0.06 | –86 |
| 80 | 10.96 | 10.91 | 15.62 | 15.50 | 4.65 | 4.59 | 0.05 | 0.12 | 0.07 | –141 |
| 7 | ||||||||||
| 40 | 14.22 | 14.25 | 16.79 | 16.79 | 2.57 | 2.53 | –0.03 | 0.01 | 0.04 | –2056 |
| 60 | 14.44 | 14.21 | 19.64 | 19.31 | 5.20 | 5.10 | 0.23 | 0.33 | 0.10 | –57 |
| 80 | 13.84 | 13.66 | 19.41 | 19.14 | 5.57 | 5.48 | 0.18 | 0.28 | 0.09 | –67 |
| 8 | ||||||||||
| 40 | 20.17 | 21.05 | 21.39 | 21.97 | 1.22 | 0.92 | –0.88 | –0.58 | 0.30 | 197 |
| 60 | 21.40 | 22.00 | 26.69 | 26.85 | 5.28 | 4.85 | –0.60 | –0.16 | 0.43 | 864 |
| 80 | 16.56 | 16.38 | 22.48 | 21.95 | 5.92 | 5.57 | 0.18 | 0.53 | 0.35 | –168 |
| 9 | ||||||||||
| 40 | 11.55 | 10.54 | 9.67 | 9.14 | –1.87 | –1.40 | –1.01 | –0.54 | 0.47 | 306 |
| 60 | 8.17 | 6.74 | 6.78 | 5.89 | –1.40 | –0.84 | –1.44 | –0.88 | 0.56 | 230 |
| 80 | 6.75 | 4.95 | 4.36 | 3.17 | –2.39 | –1.78 | –1.80 | –1.19 | 0.61 | 194 |
| 10 | ||||||||||
| 40 | 14.08 | 13.65 | 11.49 | 11.08 | –2.59 | –2.58 | –0.42 | –0.41 | 0.01 | 42 |
| 60 | 10.41 | 9.43 | 8.34 | 7.44 | –2.07 | –1.99 | –0.98 | –0.90 | 0.08 | 61 |
| 80 | 8.26 | 7.00 | 5.33 | 4.20 | –2.93 | –2.81 | –1.26 | –1.14 | 0.12 | 68 |
| 11 | ||||||||||
| 40 | 12.84 | 12.83 | 10.70 | 10.70 | –2.14 | –2.13 | –0.007 | –0.003 | 0.004 | 477 |
| 60 | 7.20 | 7.10 | 6.37 | 6.29 | –0.83 | –0.81 | –0.10 | –0.08 | 0.02 | 110 |
| 80 | 4.72 | 4.62 | 3.42 | 3.34 | –1.30 | –1.28 | –0.10 | –0.08 | 0.02 | 97 |
Для энантиомеров кислот (1–5, 9–11) монотонное снижение |ΔH0| сопровождается восходящей ветвью зависимости k' от концентрации этанола в элюенте в диапазоне 60–80 об. % (рис. 1а), что связано с реализацией ионных взаимодействий, которые, вероятно, являются термодинамически более выгодными, чем гидрофобные взаимодействия. Известно, что при адсорбции из водно-органических растворов взаимодействие адсорбата с поверхностью адсорбента и сольватация молекул адсорбата в элюенте, как правило, вносят сопоставимый вклад в суммарный тепловой эффект [29, 30]. Так, увеличение содержания этанола в элюенте способствует обогащению сольватных оболочек адсорбатов и адсорбционного слоя его молекулами [31, 32]. Поэтому уменьшение абсолютной величины |ΔH0| для энантиомеров ГК с ростом концентрации этанола в элюенте, вероятно, обусловлено увеличением энергетических затрат на разрушение сольватных оболочек молекул адсорбатов и вытеснение молекул этанола молекулами адсорбатов из поверхностного слоя в объемный раствор в силу конкурентного характера адсорбции из водно-спиртовых растворов.
Уменьшение абсолютной величины энтропийного терма с увеличением концентрации этанола в элюенте является дополнительным аргументом в пользу данного предположения (табл. 2). То есть при увеличении степени обогащения поверхностного слоя молекулами этанола, адсорбция энантиомера приводит к вытеснению в объемный раствор большего числа молекул модификатора, и, следовательно, сообщению молекулам элюента большего числа степеней свободы [31, 32]. Последнее, вероятно, компенсирует снижение числа степеней свободы молекулы энантиомера при переходе в адсорбированное состояние.
Зависимости теплоты и энтропии адсорбции от концентрации этанола в ПФ для энантиомеров эфиров 6–8 имеют максимум при 60 об. % этанола (табл. 2), что может свидетельствовать об изменении механизма удерживания. Вероятно, адсорбция 6–8 протекает не по конкурентному механизму. То есть адсорбция энантиомеров эфиров на модифицированной молекулами этанола поверхности хирального адсорбента не приводит к вытеснению молекул модификатора в объемный раствор и энергия взаимодействия адсорбционных центров с адсорбатом увеличивается. Как следствие, отсутствуют энергетические затраты, связанные с преодолением взаимодействия органический модификатор-адсорбент [32]. В результате чего число степеней свободы в системе уменьшается, приводя к увеличению энтропийного терма (табл. 2).
Взаимосвязь термодинамики адсорбции с удерживанием и селективностью. Удерживание адсорбатов связано с изменением стандартной энергии Гиббса (ΔG0) при адсорбции:
Вклад энтропии в свободную энергию адсорбции ГК 1–5, 9–11 больше половины величины ΔH0, а в случае эфиров 6–8 энтропийный терм превышает энтальпийный (табл. 2), указывая на рост разупорядоченности в системе и образование нестабильного поверхностного комплекса адсорбат–адсорбент. Поэтому стерические факторы оказывают большее влияние на удерживание энантиомеров исследуемых соединений, чем энергетические изменения. О значимости стерического фактора в энантиоразделении свидетельствует тот факт, что в элюентах с концентрацией этанола 60 и 80 об. % абсолютные значения теплоты и энтропии адсорбции слабее удерживаемого S-энантиомера эфиров 6–8 превышают значения |ΔH0| и |TΔS0| для сильнее удерживаемого R-энантиомера, при этом порядок элюирования не изменяется [30]. Максимальными значениями |ΔH0| и |TΔS0| обладают энантиомеры эфира 8, содержащего два ароматических фрагмента в молекуле, что свидетельствует об определяющей роли структурных преобразований в адсорбционной системе, связанных с реализацией дополнительных π–π-взаимодействий адсорбат–адсорбент.
Разности термодинамических характеристик адсорбции оптических антиподов (ΔΔH0, ΔΔS0 и ΔΔG0) связаны с селективностью ХНФ следующим образом :
(2)
$\begin{gathered} - RT\ln \alpha = \Delta \Delta {{G}^{0}} = \Delta G_{{\text{R}}}^{0} - \Delta G_{{\text{S}}}^{0} = \\ = \Delta \Delta {{H}^{0}} - T\Delta \Delta {{S}^{0}}. \\ \end{gathered} $При всех исследуемых условиях энтальпийный терм меньше нуля и определяет энантиоразделение кислот 1–5, 9–11, так как значительно превышает энтропийный вклад, который корректирует α в сторону уменьшения. Для эфиров 6–8 с ростом концентрации этанола в элюенте энтропийный терм начинает определять энантиоразделение, поскольку для них ΔΔН0 > 0 (табл. 2).
Термодинамические характеристики уравнения (2) позволяют определить изоэнантиоселективную температуру (Tiso), при которой энантиомеры не разделяются. Выше значения Tiso порядок элюирования энантиомеров может измениться на обратный [33]:
Tiso для всех рассматриваемых соединений (табл. 2) находится вне рабочего диапазона температуры (22–50°C). Исключением является кислота 10, для которой Tiso равна 42°C в элюенте с концентрацией этанола 40 об. % (табл. 2). Действительно, экспериментальные данные показывают, что в элюенте с концентрацией этанола 40 об. % при температуре 30°C первым элюируется R-энантиомер, при 40°C – S-энантиомер (данные не приводятся).Для кислот 1–5, 9, 11 значения Tiso находятся выше исследуемого температурного диапазона (табл. 2), указывая на то, что механизм энантиоразделения не изменяется в исследуемом температурном интервале и более низкая температура предпочтительна для лучшего энантиоразделения [34]. Обратное наблюдается для эфиров 6–8.
Энтальпийно-энтропийная компенсация. Известно, что в ряду родственных соединений может наблюдаться явление энтальпийно-энтропийной компенсации (компенсационного эффекта (КЭ)) [35, 36], что выражается линейной зависимостью между термодинамическими характеристиками адсорбции, указывая на сходство механизма адсорбции в группе соединений:
где β – компенсационная температура, $\Delta G_{\beta }^{0}$ – энергия Гиббса адсорбции при температуре β.Показано [36–38], что статистический эффект, связанный с погрешностями определения величин ΔH0 и ΔS0 по уравнению (1), может вести к проявлению ложного КЭ [19]. Более надежный метод поиска экстратермодинамических корреляций заключается в расчете характеристик ΔH≠ и ΔG≠ с помощью уравнения (3) и проверки линейности зависимости ΔH≠ от ΔG≠ [38]:
(3)
$\ln \frac{{k{\kern 1pt} '}}{\varphi } = - \frac{{\Delta H_{{}}^{ \ne }}}{R}\left( {\frac{1}{T} - \frac{1}{{{{T}_{{hm}}}}}} \right) - \frac{{\Delta G_{{}}^{ \ne }}}{{R{{T}_{{hm}}}}}$,Уравнение КЭ в данном случае имеет вид:
При соблюдении условия линейности угловой коэффициент и свободный член этой зависимости равны γ = 1/(1 – Thm/β) и $\Delta G_{\beta }^{0}$(1 – γ), соответственно [38].
На рис. 2 представлены термодинамические зависимости в координатах ΔH0 – ΔS0 (рис. 2а, б) и ΔH≠ – $\Delta G_{{}}^{ \ne }$(2 в, г) при различной концентрации этанола в элюенте. КЭ наблюдается для S- и R‑энантиомеров трех отдельных групп адсорбатов (табл. 3). Эфиры миндальной кислоты 6–8 составляют отдельную группу III, кислоты образуют две компенсационные серии I и II, состав которых различен для S- и R-энантиомеров. Для слабее удерживаемых S-энантиомеров точки, соответствующие кислотам 3, 4, 9–11 образуют компенсационную серию II, на некотором удалении от которой находятся точки кислот 1, 2 и 5. S-Энантиомеры последних образуют компенсационную серию I с коэффициентом корреляции r2 ≥ ≥ 0.90 (рис. 2а, в, табл. 3). Однако, ранее нами [19, 20] выявлено различие в механизме адсорбции S-энантиомеров кислот 1 и 5, связанное с особенностями их пространственного строения. Поэтому в данном случае мы не можем утверждать о существовании КЭ для кислот 1, 2 и 5. Для более конкретных выводов необходимо исследование адсорбции энантиомеров большего числа производных ГК.
Рис. 2.
Компенсационные зависимости для энантиомеров гидроксикислот и их производных в координатах ΔH0 – ΔS0 (а, б) и ΔH≠ – ΔG≠ (в, г) при содержании этанола в элюенте 40 об. % (1), 60 об. % (2) и 80 об. % (3); S-энантиомеры отмечены темными символами, R-энантиомеры – светлыми символами. Римскими цифрами отмечены компенсационные группы соединений.

Таблица 3.
Значения компенсационной температуры (β и β'), коэффициентов корреляции (r2) и достигнутого уровня значимости гипотезы β = Thm(γ) для компенсационных зависимостей, построенных в координатах ΔH0 – ΔS0 и ΔH≠ – $\Delta G_{{}}^{ \ne }$
| [Этанол], об. % | Группа | Соединения | ΔH0(ΔS0) | ΔH≠(ΔG≠) | ||||
|---|---|---|---|---|---|---|---|---|
| β, K | r2 | γ | β′, K | r2 | γ | |||
| S-энантиомеры | ||||||||
| 40 | I | 1, 2, 5 | 471 | 0.9657 | 0.1185 | 519 | 0.9053 | 0.1991 |
| II | 3, 4, 9, 10, 11 | 519 | 0.9622 | 0.0032 | 569 | 0.9347 | 0.0072 | |
| III | 6, 7, 8 | 383 | 0.9974 | 0.0326 | 388 | 0.9589 | 0.1300 | |
| 60 | I | 1, 2, 5 | 456 | 0.9836 | 0.0819 | 479 | 0.9384 | 0.1597 |
| II | 3, 4, 9, 10, 11 | 530 | 0.9918 | 3.17 × 10–4 | 539 | 0.9455 | 0.0055 | |
| III | 6, 7, 8 | 287 | 0.9966 | 0.0374 | 274 | 0.5565 | 0.4639 | |
| 80 | I | 1, 2, 5 | 401 | 0.9951 | 0.0446 | 409 | 0.9507 | 0.1426 |
| II | 3, 4, 9, 10, 11 | 551 | 0.9953 | 1.35 × 10–4 | 541 | 0.9355 | 0.0071 | |
| III | 6, 7, 8 | 251 | 0.9980 | 0.0284 | 249 | 0.9443 | 0.1516 | |
| R-энантиомеры | ||||||||
| 40 | I | 1, 2, 3, 4, 5 | 362 | 0.9984 | 2.63 × 10–5 | 365 | 0.9513 | 0.0046 |
| II | 9, 10, 11 | 483 | 0.9947 | 0.0465 | 490 | 0.9840 | 0.0806 | |
| III | 6, 7, 8 | 391 | 0.9980 | 0.0286 | 395 | 0.9736 | 0.1040 | |
| 60 | I | 1, 2, 3, 4, 5 | 356 | 0.9973 | 5.88 × 10–5 | 363 | 0.9017 | 0.0135 |
| II | 9, 10, 11 | 558 | 0.9839 | 0.0809 | 786 | 0.9665 | 0.1172 | |
| III | 6, 7, 8 | 300 | 0.9961 | 0.0400 | – | 0.1205 | – | |
| 80 | I | 1, 2, 3, 4, 5 | 348 | 0.9944 | 1.78 × 10–4 | 365 | 0.7682 | 0.0511 |
| II | 9, 10, 11 | 697 | 0.9247 | 0.1769 | –2836 | 0.9929 | 0.0538 | |
| III | 6, 7, 8 | 260 | 0.9949 | 0.0454 | 253 | 0.8189 | 0.2799 | |
Отдельную компенсационную серию II образуют точки, соответствующие R-энантиомерам кислот 9–11, удерживаемых слабее по сравнению с их оптическими антиподами (рис. 2б, г), что свидетельствует о различии механизма адсорбции ГК, имеющих противоположный порядок элюирования энантиомеров.
Наблюдаемые компенсационные закономерности, на наш взгляд, следует отнести к истинному КЭ, поскольку линейные корреляции (r2 > 0.92) для S- и R-энантиомеров трех групп соединений в координатах ΔH0 – ΔS0 (табл. 3, рис. 2а, б) при всех экспериментальных условиях подтверждаются наличием линейных корреляций (в большинстве случаев r2 > 0.90) между величинами ΔH≠ и $\Delta G_{{}}^{ \ne }$ (табл. 3, рис. 2в, г). Достоверность полученных экспериментальных данных подтверждает удовлетворительная сходимость значений компенсационной температуры β и β', рассчитанных двумя методами (табл. 3) [37].
К доказательству истинности КЭ также относится неравенство компенсационной температуры и среднегармонической температуры (Thm) исследуемого температурного интервала (308.14 К), что характеризуется величиной достигнутого уровня значимости нуль-гипотезы β = Thm(γ) (табл. 3) [36]. Чем меньше эта величина, тем меньше вероятность правильности нуль-гипотезы β = Thm. Условие неравенства компенсационной температуры температуре Thm лучше соблюдается для ряда S-энантиомеров II группы кислот (3, 4, 9–11) (рис. 2а, в) и для R-энантиомеров I группы кислот (1–5) (рис. 2б, г). Условие β ≠ Thm определенно не соблюдается для S- и R-энантиомеров эфиров 6–8 в элюенте с концентрацией этанола 60 об. % (табл. 3).
Существенное различие значений компенсационной температуры для энантиомеров группы эфиров (III) и групп кислот (I и II) подтверждает качественное различие механизма адсорбции нейтральных и ионогенных соединений [39]. Кроме того, значения компенсационной температуры различаются для R-энантиомеров I и II групп кислот 1–5 и 9–11 соответственно (табл. 3), подтверждая выдвинутое выше предположение о различии механизма адсорбции кислот, имеющих обратный порядок элюирования энантиомеров.
Таким образом, в работе получены новые данные об особенностях разделения, удерживания и термодинамики адсорбции энантиомеров ароматических ГК и их производных на ХНФ “Nautilus-E” с привитым антибиотиком эремомицином из водно-этанольных элюентов. Показано, что хроматографическое поведение и термодинамика адсорбции ГК и их производных зависит от природы заместителей в функциональных группах и при ХЦ молекул адсорбатов. Выявлено, что этерификация карбоксильной группы ГК и наличие у ХЦ заместителей, создающих стерические затруднения, снижают энантиоселективность разделения, а дополнительный вклад π–π-взаимодействий и наличие группы –CH2 между ароматическим кольцом и ХЦ в структуре молекулы способствуют росту селективности. Установлено, что α- и β-ГК имеют противоположный порядок элюирования энантиомеров. Характер зависимостей удерживания и термодинамических величин адсорбции энантиомеров исследуемых ГК и их производных указывает на изменение механизма адсорбции при увеличении концентрации этанола в элюенте, вследствие изменения состава поверхностного слоя ХНФ. Установлено, что стерические факторы оказывают большее влияние на удерживание энантиомеров нейтральных соединений, чем энергетические изменения, в то время как адсорбция энантиомеров ионогенных соединений управляется энтальпийным термом. Статистический анализ термодинамических данных позволил выявить различие в механизме адсорбции энантиомеров ионогенных и нейтральных соединений, а также энантиомеров ГК, имеющих противоположный порядок элюирования S- и R-энантиомеров.
Исследования выполнены при финансовой поддержке Российского фонда фундаментальных исследований (код проекта № 18-03-00053-А).
Список литературы
Draelos Z.D. // Dermatol. Ther. 2000. V. 13. P. 154.
Lamberth C., Jeanguenat A., Cederbaum F. et al. // Bioorg. Med. Chem. 2008. V. 16. P. 1531.
Zheng Z., Sheng B., Gao C. et al. // Sci. Rep. 2013. V. 3. P. 3401.
Coppola G.M., Schuster H.F. α-Hydroxy Acids in Enantioselective Synthesis. Wiley–VCH. Weinheim, 1997. P. 494.
Pişkin E. // J. Biomater. Sci. Polym. Ed. 1995. V. 6. P. 775.
Kacprzak K., Maier N., Lindner W. // J. Sep. Sci. 2010. V. 33. P. 2590.
Choi W.J., Lee K.Y., Kang S.H. et al. // Sep. Purif. Technol. 2007. V. 53. P. 178.
Yadav G.D., Sivakumar P. // Biochem. Eng. J. 2004. V. 19. P. 101.
Sládková V., Dammer O., Sedmak G. et al. // Crystals. 2017. V. 7. P. 13.
Armstrong D.W., Tang Y., Chen S. et al. // Anal. Chem. 1994. V. 66. P. 1473.
Berthod A., He B.L., Beesley T.E. // J. Chromatogr. A. 2004. V. 1060. P. 205.
Кузнецов М.А., Нестеренко П.Н., Васияров Г.Г., Староверов С.М. // Журн. аналит. химии. 2008. Т. 63. № 1. С. 64.
Шаповалова Е.Н., Федорова И.А., Припорова А.А. и др. // Аналитика и контроль. 2016. Т. 20. № 2. С. 168.
Hui F., Ekborg-Ott K.H., Armstrong D.W. // J. Chromatogr. A. 2001. V. 906. P. 91.
Staroverov S.M., Kuznetsov M.A., Nesterenko P.N. et al. // J. Chromatogr. A. 2006. V. 1108. P. 263.
Федорова И.А., Шаповалова Е.Н., Староверов С.М., Шпигун О.А. // Сорбц. хромат. процессы. 2015. Т. 15. № 6. С. 769.
Решетова Е.Н., Аснин Л.Д. // Журн. физ. химии. 2009. Т. 83. № 4. С. 643.
Решетова Е.Н., Аснин Л.Д. // Там же. 2011. Т. 85. № 8. С. 1552.
Reshetova E. // J. Liq. Chromatogr. Relat. Technol. 2016. V. 39. P. 145.
Блинов А.С., Решетова Е.Н. // Журн. физ. химии. 2014. Т. 88. № 10. С. 1593.
Ali I., Kumerer K., Aboul-Enein H.Y. // Chromatographia. 2006. V. 63. P. 295.
Nikitina Y.K., Ali I., Asnin L.D. // J. Chromatogr. A. 2014. V. 1363. P. 71.
Aneja R., Luthra P.M., Ahuja S. // Chirality. 2010. V. 22. P. 479.
Berthod A., Liu Y., Bagwill C., Armstrong D.W. // J. Chromatogr. A. 1996. V. 731. P. 123.
Jandera P., Škavrada M., Klemmová K. et al. // Ibid. 2001. V. 917. P. 123.
Beesley T.E., Lee J.T. // J. Liq. Chromatogr. Relat. Technol. 2009. V. 32. P. 1733.
Chester T.L., Coym J.W. // J. Chromatogr. A. 2003. V. 1003. P. 101.
Fornstedt T. // Ibid. 2010. V. 1217. P. 792.
Аснин Л.Д., Качмарски К., Решетова Е.Н. // Изв. Акад. наук, сер. Хим. 2009. № 8. С. 1678.
Asnin L., Sharma K., Park S.W. // J. Sep. Sci. 2011. V. 34. P. 3136.
Сайфутдинов Б.Р., Пимерзин А.А. // Журн. физ. химии. 2013. Т. 87. № 12. С. 2047.
Сайфутдинов Б.Р., Курбатова С.В., Емельянова Н.С. // Там же. 2010. Т. 84. № 4. С. 760.
Rojkovičová T., Lehotay J., Meričko D. et al. // J. Liq. Chromatogr. Relat. Technol. 2004. V. 27. № 16. P. 2477.
Rojkovičová T., Lehotay J., Armstrong D. W., Čižmárik J. // Ibid. 2004. V. 27. № 20. P. 3213.
Vailaya A., Horvath C. // J. Phys. Chem. B. 1998. V. 102. P. 701.
Krug R.R., Hunter W.G., Grieger R.A. // J. Phys. Chem. 1976. V. 80. P. 2335.
Miyabe K. // Anal. Sci. 2009. V. 25. P. 219.
Krug R.R., Hunter W.G., Grieger R.A. // J. Phys. Chem. 1976. V. 80. P. 2341.
Ranatunga R., Vitha M.F., Carr P.W. // J. Chromatogr. A. 2002. V. 946. P. 47.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии