

Журнал физической химии, 2019, T. 93, № 7, стр. 992-1001
Анализ динамики взаимодействия гуминовых веществ угля с металлическим железом
Д. А. Панкратов a, *, М. М. Анучина a, А. И. Константинов a, И. В. Перминова a
a Московский государственный университет имени М.В. Ломоносова
Москва, Россия
* E-mail: pankratov@radio.chem.msu.ru
Поступила в редакцию 22.09.2018
После доработки 22.09.2018
Принята к публикации 15.10.2018
Аннотация
Представлен анализ динамики взаимодействия металлического железа с водными растворами трех препаратов от различных производителей гуминовых веществ угля. Гуминовые вещества охарактеризованы методами элементного состава, ЯМР-спектроскопии, эксклюзионной хроматографии, электронной спектроскопии поглощения и химического анализа. Выявлены различия изученных гуминовых веществ в содержании ароматических фрагментов, степени их замещенности, содержании карбоксильных групп и степени окисленности. Показано, что независимо от предыстории гуминового вещества в основе взаимодействий их растворов с железом лежит одинаковый набор последовательных реакций, который на основании данных об изменении легко контролируемых параметров (окислительно-восстановительный потенциал, рН и оптическая плотность растворов) может быть разделен на три основных этапа взаимодействия. Эти этапы взаимодействия качественно описаны в координатах диаграммы Пурбе. Специфика структурно-группового состава гуминовых веществ обусловливает различное время протекания соответствующих этапов взаимодействия. По результатам анализа главных компонентов предложена методика описания сложной последовательности реакций между гуминовыми веществами и железом в координатах двухфакторной плоскости, что позволяет однозначно отнести экспериментально регистрируемые параметры реакционной системы к конкретному этапу взаимодействия металлического железа и водных растворов гуминовых веществ, независимо от специфических свойств последних.
Коррозия конструкционных железосодержащих материалов является не только причиной разрушения механизмов и конструкций, но и источником поступления в окружающую среду железа. В зависимости от места и способа эксплуатации металлоконструкций различают атмосферную, почвенную, морскую, биологическую, контактную, щелевую, блуждающими или внешними токами и другие виды коррозии. Почвенная коррозия [1] считается настолько же разрушительной, как и морская [2]. Для металлов и сплавов почва является агрессивной средой вследствие разнообразия содержащихся в ней биологических организмов [3], высокой степени аэрации при повышенном влагосодержании и наличия большого количества химических соединений органической и неорганической природы, активно участвующих в коррозионных процессах [1]. Один из типов таких соединений – гуминовые вещества (ГВ) – природные макромолекулярные полиэлектролиты, состоящие из сотен органических компонентов, замещенных карбоксильными, фенольными, гидроксильными, карбонильными и метоксильными группами [4]. Они не только непосредственно вступают с металлами [5, 6] в кислотно-основные [7], окислительно-восстановительные реакции [8] и реакции комплексообразования [9], но и выступают в качестве поверхностно-активного реагента, проявляя диспергирующий эффект [10] и способствуя транспорту частиц продуктов коррозии [11]. Очевидно, что строгое описание кинетики коррозии металла в присутствии ГВ становится трудноразрешимой задачей [12] вследствие множественности протекающих процессов и вариативности в составе и строении ГВ.
Ранее [13] нами было показано, что взаимодействие металлического железа с водными растворами ГВ – многостадийный процесс, развивающийся по нескольким путям одновременно, в результате которого образуются стабилизированные ГВ суспензии наноразмерного смешанного оксида железа (II, III), имеющие перспективы практического применения для агрохимических целей [14]. При этом ГВ выступают в качестве многофункционального реагента, участвующего в коррозионном процессе как кислотный и окислительный реагент, способствующий переводу железа в раствор, как лиганд для ионного железа в растворе, как поверхностно-активное вещество (ПАВ), стабилизирующее наноразмерные частицы в растворе, как адсорбат, влияющий на морфологические свойства железосодержащих продуктов реакции. Поэтому можно было бы ожидать, что механизмы реакций взаимодействия металлического железа с водными растворами ГВ, их набор, последовательность и кинетика, помимо прочего, должны зависеть от их структурно-групповых свойств. В работе [15] на примере трех препаратов ГВ угольного происхождения различных производителей показано, что характер изменений динамических параметров, описывающих взаимодействие ГВ с металлическим железом, в целом не зависит от предыстории ГВ, а основные отличия наблюдаются в скорости протекания отдельных этапов указанного взаимодействия.
В настоящей работе проводится анализ зависимости динамики взаимодействия металлического железа с водными растворами ГВ от их структурно-групповых свойств и предлагается методика описания этого взаимодействия на основании легко получаемых экспериментальных данных, которая позволяет однозначно выделить его отдельные этапы, и как результат прогнозировать развитие систем “Fe – ГВ” для ГВ различного происхождения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе применяли металлическое железо в форме порошка – “Восстановленное железо 97%” (Reanal, Венгрия). Перед экспериментами его обезжиривали 98%-ным этиловым спиртом, промывали 0.01 н. раствором HCl и 4 раза водой до нейтрального значения рН по лакмусу.
Образцы ГВ угольного происхождения готовили из продуктов, производимых в промышленных масштабах и реализуемых в розницу. Так, “образец 1” готовили из “Гумата калия Сахалинский” (ООО “Биомир-2000”, г. Москва, Россия), получаемого из верхних высокоокисленных слоев бурого угля Солнцевского месторождения острова Сахалин. При этом коммерческий препарат предварительно очищали от нерастворимых примесей неорганического происхождения путем циклического повторения операций растворения, декантации, центрифугирования и упаривания растворов с последовательно уменьшающимися в циклах концентрациями препарата, аналогично [16]. После указанных операций массовая доля золы в “образце 1” снизилась с 32.0% для исходного коммерческого продукта “Гумат калия Сахалинский” до 27.1%.
Препарат “Natural Humic Acids” (Life Force, г. Саратов, Россия), производимый посредством низкотемпературной физико-механической обработки лигнитов и суббитуминозных углей, преимущественно представляет собой нерастворимые в воде гуминовые кислоты, поэтому методом щелочной экстракции его переводили в водорастворимую форму (далее “образец 2”) [15]. При этом выход растворимой части ГВ из коммерческого препарата составил 40%.
Коммерческий продукт “PowHumus” (Humintech, г. Дюссельдорф, Германия), выделяемый с помощью щелочной экстракции из разновидности окисленного бурого угля – леонардита, полностью растворяется в воде без признаков выделения осадков. Поэтому его непосредственно использовали в качестве “образца 3” без какой-либо дополнительной обработки.
Содержание C, H и N в образцах определяли в лаборатории микроанализа ИНЭОС им. А.Н. Несмеянова РАН. Элементный состав ГВ пересчитывали на беззольное органическое вещество. Содержание кислорода рассчитывали по разности: [O],% = 100 – ([C] + [H] + [N]).
Исследование структурно-группового состава ГВ осуществляли методом количественной спектроскопии 13С ЯМР на ЯМР-спектрометре “Avance 400” компании “Bruker” с рабочей частотой для ядер 13С 100 МГц. Для этого навеску ГВ ~40 мг растворяли в 0.6 мл 0.3 М NaOD/D2O и помещали в 5 мм ампулу для ЯМР. Количественные спектры регистрировали с применением импульсной последовательности CPMG, при этом широкополосная развязка от протонов во время регистрации сигнала спада свободной индукции выключали для исключения ядерного эффекта Оверхаузера (импульсная техника INVGATE). Для полной релаксации всех ядер 13С длительность релаксационной задержки составляла 7.8 с, и общее время одного эксперимента ЯМР составляло ~10 ч. Структурно-групповой состав ГВ определяли интегрированием соответствующих спектральных областей согласно [17].
Молекулярно-массовое распределение ГВ определяли с помощью эксклюзионной хроматографии. Фракционирование проводили на хроматографической системе Abimed, состоящей из ВЭЖХ-насоса, автосамплера, стеклянной колонки (∅25 мм, L = 20 см), спектрофотометрического УФ-детектора и регистрирующего компьютера. Колонку заполняли гелем “Toyopearl” TSK HW-55S (Toso–Haas, Япония) с диапазоном фракционирования 1–200 кДa по полидекстранам. В качестве подвижной фазы использовали фосфатный буфер (0.03 М, рН 6.8). Условия анализа аналогичны описанным в [18]. Для калибровки использовали натриевые соли полистиролсульфоновых кислот (Polymer Standard Service, Германия). На основе полученных хроматограмм и калибровочных кривых находили значения средневесовых молекулярных масс.
С помощью спектрофотометра 6715 UV/Vis. (JENWAY, Великобритания) получали спектры поглощения в УФ- и видимой областях спектра в диапазоне 190–1100 нм, с применением кварцевых кювет l = 1 см и воды в качестве раствора сравнения. Приводимые в работе значения Еλ (экстинкция на длине волны λ, нм) получали умножением регистрируемой оптической плотности разбавленной пробы на кратность ее разбавления. Измерения pH и Eredox производили на лабораторном иономере И-160МИ (Измерительная техника, Россия).
Показатель перманганатной окисляемости (ППО) в исходных препаратах ГВ определяли по модифицированному нами методу Кубеля [19]. Для этого к анализируемой пробе, содержащей 250 мг/л ГВ, объемом 1 мл последовательно при перемешивании добавили 9 мл воды, 2.5 мл раствора серной кислоты (1 : 3) и 10 мл 0.01 моль-экв/л раствора перманганата калия. Полученный раствор кипятили в закрытой часовым стеклом колбе в течение 10 мин. Затем к горячему раствору добавили 10 мл 0.01 моль-экв/л раствора щавелевой кислоты и через 10 мин после охлаждения до комнатной температуры титровали 0.01 моль-экв/л раствором перманганата калия. Определение ППО для каждой пробы проводили 3 раза. Значение ППО по Кубелю в мг О/г ГВ рассчитывали с учетом холостого опыта по формуле:
(1)
$\begin{gathered} {\text{П П О }} = \frac{{({{V}_{1}} - {{V}_{0}})K \times 0.01 \times 8 \times 1000 \times 4}}{V}\; \times \\ \times \;\frac{{100}}{{1000\omega \% }} = \frac{{({{V}_{1}} - {{V}_{0}})K \times 32000}}{{V(100 - \omega \% )}}, \\ \end{gathered} $Все работы проводили с использованием тридистиллированной воды. При проведении аналитических работ использовали реактивы и реагенты марки “ос.ч.”, либо стандартные фиксаналы.
Эксперименты по изучению динамики взаимодействия растворов ГВ с металлическим железом проводили по следующей методике. Из полученных как указано выше сухих водорастворимых порошков приготовили 250 мг/л растворы ГВ. Раствор ГВ объемом 1 л и 10 г металлического железа герметично закрывали в полиэтиленовом сосуде, и перемешивали путем полного переворачивания с частотой 45 об./мин. Через известные интервалы времени (20–50 ч) перемешивание останавливали на 1 ч для осаждения крупной взвеси (при открытой крышке) и на глубине 1 см с помощью дозатора отбирали пробы по 10 мл для контроля рН, Eredox и Е254. Затем контейнеры вновь герметично закрывали и продолжали перемешивание. Эксперименты продолжали до полного обесцвечивания надосадочной жидкости. В случае образца 1 он длился в течение 265 ч, образца 2 – 311 ч, образца 3 – 412 ч. Эксперименты проводили при комнатной температуре без термостатирования. Для образца 1 средняя температура составила 22°С (эксперимент проводили отдельно), для образцов 2 и 3 – 19°С (эксперименты проводили одновременно).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В работе рассматривали взаимодействие трех образцов ГВ с металлическим железом. Несмотря на схожую природу изучаемых образцов ГВ, их физико-химические свойства могут различаться вследствие различий в методиках их извлечения из ископаемого сырья, в предыстории этого сырья, мест его добычи.
По данным элементного анализа (табл. 1), образец 3, несмотря на то, что он предварительно не очищался от минеральных и нерастворимых примесей, заметно выделялся низким содержанием несгораемого остатка (табл. 1). Кроме того, образец 3 отличался от других заметно более низким значением отношения Н/С, отражающем его высокую степень ненасыщенности. Образец 1 отличался от других более низким содержанием углерода и высокими – кислорода и азота. Как результат, данный образец характеризовался максимальным значением отношения О/С, указывающим на большую обогащенность данного препарата кислородсодержащими функциональными группами. Образец 2, в свою очередь, по степени обогащенности кислородом близок к образцу 3, а по степени ненасыщенности – к образцу 1.
Таблица 1.
Данные по элементному, структурно-групповому и молекулярно-массовому составу изученных препаратов ГВ 1–3
| Свойства | 1 | 2 | 3 | |||||
|---|---|---|---|---|---|---|---|---|
| Массовая доля золы (ω, %) | 27.1 | 26.4 | 18.7 | |||||
| Содержание элементов в пересчете на беззольную пробу, мас. % |
C | 48.11 | 55.96 | 57.80 | ||||
| H | 4.86 | 5.91 | 4.57 | |||||
| N | 1.77 | 1.40 | 1.56 | |||||
| O | 45.27 | 36.73 | 36.07 | |||||
| Атомные соотношения | Н/С | 1.21 | 1.27 | 0.95 | ||||
| О/С | 0.71 | 0.49 | 0.47 | |||||
| Распределение по структурным фрагментам, % от общего С |
C=O | 6.0 | 5.4 | 7.0 | ||||
| COO | 19.4 | 10.8 | 14.2 | |||||
| ΣCAr | CArO | 52.3 | 10.5 | 52.1 | 12.2 | 51.6 | 10.5 | |
| CAr | 41.8 | 39.9 | 41.1 | |||||
| ΣCAlk | OCO | 22.3 | 4.5 | 31.7 | 8.1 | 27.3 | 5.4 | |
| СAlkO | 6.4 | 10.7 | 6.5 | |||||
| CHn | 11.4 | 12.9 | 15.4 | |||||
| Средневесовая молекулярная масса, кДa | 12.7 | 14.7 | 12.6 | |||||
Спектры 13С ЯМР исследованных образцов (рис. 1) типичны для ГВ угля. Для них характерен интенсивный широкополосный сигнал в интервале 100–165 м.д., соответствующий углероду ароматических структур, и относительно малоинтенсивный сигнал алифатических структур (0–100 м.д.), в особенности – алкоксигрупп (50–100 м.д.). Также спектры характеризуются интенсивными сигналами в области 165–185 м.д., соответствующими углероду карбоксильных и сложноэфирных групп. Количественные характеристики структурно-группового состава исследованных веществ, полученные интегрированием спектров 13С ЯМР, приведены в табл. 1.

Спектры поглощения исходных растворов ГВ в изученном диапазоне длин волн от 200 до 800 нм имеют вид протяженной ниспадающей кривой без отчетливо выраженных полос поглощения [20] (рис. 2). Оптические свойства растворов ГВ определяются такими структурными параметрами как молекулярная масса, относительное содержание ароматических и алифатических фрагментов в составе ГВ, а также природой, содержанием и положением в них функциональных групп [21]. Анализ оптических свойств проводится с применением численных дескрипторов. Так, сравнивая величины оптической плотности при 254 нм (Е254) для растворов ГВ с известными концентрациями или те же величины, нормированные на содержание углерода (SUVA254), можно оценить степень гумификации и ароматичности ГВ [22]. Уменьшение средних размеров молекул ГВ приводит к уменьшению поглощения высокомолекулярных фракций ГВ в длинноволновой области оптического спектра и, следовательно, к увеличению отношения E250 и Е365 (дескриптор E2/E3) [23]. Дескриптор E4/E6 (отношение Е465 и Е665) обратно пропорционален протяженности ароматической системы ГВ – степени конъюгации, и прямо пропорционален относительному содержанию в ней карбоксильных заместителей [24]. Низкая степень замещения ароматических колец, а также присутствие заместителей в виде алифатических функциональных групп служат причинами низкого значения для дескриптора EET/EBZ (отношение Е253 и Е220) [25]. Увеличивает значение EET/EBZ высокая степень замещения ароматических колец ГВ гидроксильными, карбонильными, карбоксильными или эфирными группами.
Рис. 2.
Электронные спектры поглощения разбавленных в 10 раз исходных растворов препаратов ГВ угля различных производителей (1–3) 25 мг/л (указаны положения длин волн поглощения, применяемые для расчета значений дескрипторов).
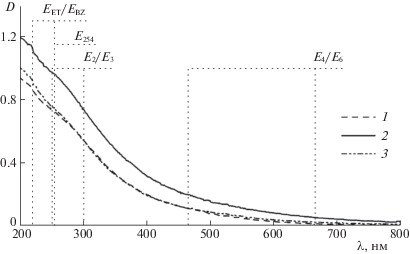
Полученные для исходных растворов ГВ дескрипторы E2/E3 различаются не существенно (табл. 2). Это указывает на то, что средние размеры молекул ГВ в препаратах также различаются незначительно, и согласуется с данными о молекулярно-массовом распределении для рассмотренных в работе образцов (табл. 1). Существенные различия наблюдаются для дескриптора E4/E6, указывающего, в том числе, на степень конъюгации ГВ. Так, для образцов 1 и 2 наблюдаются соответственно максимальное и минимальное значения дескриптора E4/E6, отвечающие наименьшей и наибольшей степени конъюгации (табл. 2). Эти данные согласуются со значениями дескриптора SUVA254, пропорционального степени ароматичности ГВ. В совокупности с данными структурно-группового анализа (табл. 1) можно заключить, что образец 1 отличается наименьшим содержанием ароматических фрагментов, а образец 2 – наибольшим. Дескриптор E4/E6 также хорошо коррелирует с данными структурно-группового анализа по относительному содержанию в образцах карбоксильных групп (%COO, табл. 1): образец 2 отличается минимальным содержанием карбоксильных групп, а образец 1 – максимальным. Одинаково низкие значения дескрипторов EET/EBZ (табл. 2) свидетельствуют о невысокой степени замещения в ароматических фрагментах для всех изученных образцов ГВ.
Таблица 2.
Свойства исходных 250 мг/л растворов ГВ 1–3
| Свойства | 1 | 2 | 3 | |
|---|---|---|---|---|
| рН | 9.56 | 8.13 | 8.90 | |
| Еredox, мВ | 188.9 | 238.0 | 154.0 | |
| ППО, мг О/г ГВ | * | 61.28 ± 0.96 | 58.40 ± 0.76 | 82.80 ± 0.76 |
| 84.0 ± 1.3 | 79.36 ± 1.03 | 101.85 ± 0.93 | ||
| Дескрипторы структуры ГВ | $Е _{{254}}^{*}$ | 7.26 | 9.65 | 7.49 |
| SUVA254, л/(м мгС) | 8.28 | 9.37 | 6.37 | |
| Е2/E3 | 2.57 | 2.26 | 2.71 | |
| E4/E6 | 9.42 | 3.74 | 4.78 | |
| EET/EBZ | 0.83 | 0.85 | 0.81 | |
Таким образом, по совокупности приведенных результатов, а также данных потенциометрического (Еredox, табл. 2) и перманганатно-метрического (ППО, табл. 2) анализов можно заключить, что из трех исследуемых препаратов ГВ угля образец 1 характеризуется самой низкой степенью конъюгации, но содержит наибольшее число карбоксильных заместителей; образец 2 отличается высокой ароматичностью, высокой степенью окисленности, но минимальным содержанием карбоксильных групп; образец 3 низкоокислен, а по ароматичности и содержанию карбоксильных групп занимает промежуточное положение.
При осуществлении взаимодействия металлического железа с водными растворами ГВ по указанной выше методике наблюдаются существенные изменения интенсивности окраски растворов. Так, в начале взаимодействия оптическая плотность исходных интенсивно-коричневых растворов еще больше повышается, вплоть до образования черных непрозрачных растворов, а затем резко падает с образованием практически бесцветных растворов. Анализ дескрипторов ГВ для этих растворов нецелесообразен, так как изменение интенсивности окраски растворов обусловлено увеличением рассеяния света на взвешенных частицах продуктов взаимодействия железа и ГВ.
Ранее в работе [13] было предположено, что взаимодействие ГВ и металлического железа осуществляется по сложному многостадийному механизму, включающему ряд последовательных и параллельных реакций:
– взаимодействие железа и воды с выделением водорода
(2)
${\text{F}}{{{\text{e}}}^{0}} + {\text{2}}{{{\text{H}}}_{2}}{\text{O}} \to {\text{F}}{{{\text{e}}}^{{2 + }}} + 2{\text{O}}{{{\text{H}}}^{--}} + {{{\text{Н }}}_{2}},$– взаимодействие гуминовых кислот и железа с выделением водорода
(3)
${\text{F}}{{{\text{e}}}^{0}} + 2{\text{HA}} \to {\text{F}}{{{\text{e}}}^{{2 + }}} + {{{\text{H}}}_{2}} + 2{{{\text{A}}}^{--}},$– окислительно-восстановительное взаимодействие ГВ и железа
(4)
${\text{F}}{{{\text{e}}}^{0}} + {\text{Г }}{{{\text{В }}}_{{{\text{Ox}}}}} \to {\text{F}}{{{\text{e}}}^{{2 + }}} + {\text{Г }}{{{\text{В }}}_{{{\text{Red}}}}},$– образование растворимых комплексов железа и ГВ
(5)
${\text{F}}{{{\text{e}}}^{{2 + }}} + {\text{Г }}{{{\text{В }}}^{n}}^{ - } \to {{[{\text{Fe(Г В )}}]}^{2}}^{{--n}},$– образование малорастворимого гидроксида железа (II)
(6)
${\text{F}}{{{\text{e}}}^{{2 + }}} + 2{\text{O}}{{{\text{H}}}^{--}} \to {\text{Fe}}{{({\text{OH}})}_{2}},$– окисление ионов железа (II) в железо (III) кислородом воздуха
(7)
$4{\text{F}}{{{\text{e}}}^{{2 + }}} + 2{{{\text{H}}}_{2}}{\text{O}} + {{{\text{О }}}_{2}} \to 4{\text{F}}{{{\text{e}}}^{{3 + }}} + 4{\text{O}}{{{\text{H}}}^{--}},$– образование нерастворимых частиц смешанного оксида железа (II, III):
(8)
${\text{F}}{{{\text{e}}}^{{2 + }}} + 2{\text{F}}{{{\text{e}}}^{{3 + }}} + 8{\text{O}}{{{\text{H}}}^{--}} \to {\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}} + 4{{{\text{H}}}_{2}}{\text{O}},$– адсорбция ГВ на частицах смешанного оксида железа (II, III)
(9)
${\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}} + {\text{Г В }} \to {\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}}{\text{@Г В }}.$После удаления из раствора всех ГВ наблюдается обычная коррозия оставшегося металлического железа:
(10)
$4{\text{Fe}} + 6{{{\text{H}}}_{2}}{\text{O}} + 3{{{\text{O}}}_{2}} = 4{\text{Fe}}{{({\text{OH}})}_{3}}.$Развитие системы “раствор ГВ–железо”, включающее реакции (2)–(10), сопровождается изменением ряда параметров, например, таких как E254, Eredox, ППО, рН, CFe, DДРС11 [13]. В настоящей работе ограничились такими легко регистрируемыми параметрами, как E254, Eredox, рН, которые, согласно [13], достаточно полно описывают динамику взаимодействий в указанной системе.
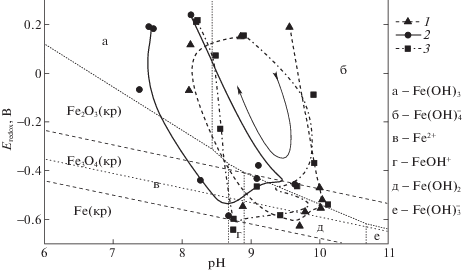
Качественно описать процесс взаимодействия металлического железа с водными растворами ГВ можно в координатах диаграммы Пурбе [26]. На рис. 3 приводятся экспериментальные данные для изученных образцов на фоне областей термодинамической устойчивости растворимых и нерастворимых соединений железа (без учета ионной силы растворов и комплексообразования соединений железа) при заданных значениях рН и окислительно-восстановительного потенциала. Несмотря на то, что данные для образца 2 немного смещены в область низких рН, характер изменений во времени параметров (рН, Eredox) для всех образцов одинаков, имеет υ-образный вид (см. стрелку на рис. 3), и может быть условно разделен на три этапа взаимодействия. На первом этапе параметры системы (рН, Eredox), первоначально находящейся в области существования соединений железа (III), изменяются в направлении уменьшения потенциала и роста рН (правая часть υ) вплоть до границы областей существования ${\text{Fe}}({\text{OH}})_{4}^{ - }$ и Fe(OH)2 (области б и д рис. 3). На втором этапе параметры системы изменяются в пределах областей существования соединений железа (II) (рис. 3 г, д) и кристаллического Fe3O4 (нижняя часть υ). Третий этап сопровождается резким изменением параметров (левая часть υ) до значений, соответствующих области устойчивости Fe(OH)3 (рис. 3а).
Рис. 3.
Диаграмма Пурбе для железа с общей концентрацией 0.01 М при 25°С и изменение Eredox и рН для растворов препаратов ГВ угля (1–3) в процессе их взаимодействия с металлическим железом. Стрелкой указано направление развития изучаемых систем. Пунктиром разделены области термодинамической стабильности кристаллических фаз соединений железа. Линия точками соответствует восстановлению воды.
На основании анализа изменений параметров E254 и Eredox, развитие изучаемых систем также можно разделить на три этапа. На первом наблюдается быстрое возрастание E254 (до точки перегиба зависимости E254 от времени, рис. 4а) и резкое уменьшение Eredox (рис. 4в). Очевидно, что в указанном интервале времени, согласно (2)–(4), в растворах накапливаются растворимые соединения железа и восстанавливаются ГВ. На втором этапе скорость возрастания E254 заметно уменьшается (от точки перегиба зависимости E254 от времени до его максимального значения (рис. 4а), а Eredox практически не изменяется (рис. 4в). Этот интервал времени соответствует накоплению устойчивых взвесей частиц малорастворимых соединений железа (II) согласно (5), (6). Третий этап сопровождается резким уменьшением E254 и одновременным увеличением Eredox до первоначального значения. На этом этапе преимущественно происходит окисление Fe2+ до Fe3+, формирование магнетита и совместное соосаждение с ГВ – реакции (7)–(9).
Рис. 4.
Изменения показателя Е254 (а), значений рН (б), окислительно-восстановительного потенциала (в), а также их нормализованных значений Е254 (г), рН (д), и окислительно-восстановительного потенциала (е) для растворов препаратов ГВ различных производителей (1–3) в процессе их взаимодействия с металлическим железом.
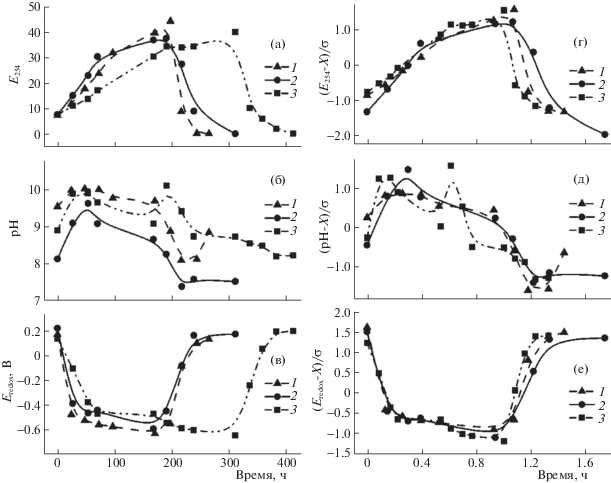
Несмотря на то, что динамика изменений значений рН растворов менее выразительна (рис. 4б), для этого параметра также можно выделить участки, соответствующие указанным выше этапам. Так, на первом этапе, в соответствии с (2), (3), наблюдается увеличение рН. На втором этапе взаимодействия постепенное уменьшение рН связано с участием функциональных групп ГВ в образовании комплексов с железом Fe2+, согласно (5), и связыванием гидроксильных ионов согласно (6). Причиной уменьшения рН на заключительном этапе является как связывание гидроксильных ионов по (8), так и вывод ГВ из раствора в результате сорбции на оксидах железа согласно (9).
Для облегчения анализа динамики изменения параметров изучаемой системы и нивелирования действия различных факторов на них (природа ГВ, температура синтеза, интервалы времени взятия проб, интенсивность перемешивания и прочие плохо контролируемые факторы) полученные экспериментальные данные были подвергнуты нормировке. Для этого экспериментальные данные, характеризующие изменения в растворах (E254, рН, Eredox), были отцентрированы и нормированы на величину стандартного отклонения. Шкалу времени нормировали относительно образца 1, опираясь на характеристичную точку, отвечающую окончанию второго и началу третьего этапов взаимодействия, и отличающуюся максимальным значением параметра E254 и минимальным значением Eredox (рис. 4 г–е). Нормировочные коэффициенты времени для образцов 2 и 3 относительно образца 1 составили 1.03 и 0.59 соответственно.
Нормировка экспериментальных данных позволяет сравнить изменения основных контролируемых параметров систем на условной шкале времени взаимодействия. Так, из полученных данных становится очевидным, что развитие изучаемых систем протекает по единой схеме, т.е. независимо от методов выделения и предыстории ГВ взаимодействия в системах “раствор ГВ – металлическое железо” протекают согласно одинаковой последовательности реакций, например, указанных выше (2)–(9). Источник ГВ оказывает влияние, судя по всему, только на время протекания реакций каждого из условных этапов взаимодействия. Так, продолжительность первого этапа взаимодействия реагентов, согласно рис. 4 г–е, составляет 0.55, 0.40 и 0.60 о.е. времени для образцов 1, 2 и 3 соответственно. Причем в абсолютной шкале времени для образцов 2 и 3, изученных в условиях параллельного эксперимента, длительности первых этапов различаются более чем в 2 раза (рис. 4 а–в). Продолжительность третьего этапа взаимодействия для указанных образцов составляет 0.3, 0.5 и 0.2 о.е. времени (рис. 4 г–е) (тогда как в абсолютных величинах длительность этого этапа близка).
Таким образом, образец 2 отличается короткой продолжительностью первого этапа взаимодействия и продленной – третьего. Причинами этого могут служить различия в структуре и составе исследуемых ГВ. Так, образец 2 по совокупности данных был охарактеризован как препарат с высокой степенью окисленности и ароматичности. Очевидно, что высокая акцепторная способность образца 2 (энтальпийный фактор), с одной стороны, и возможность переноса электронной плотности по сопряженным связям ароматических фрагментов макромолекул ГВ (энтропийный фактор), с другой стороны, создают благоприятные условия для взаимодействия ГВ и железа согласно (4). А минимальная окисленность образца 3 уменьшает скорость взаимодействия ГВ с железом по (4) на первом этапе. Минимальное содержание в составе препарата 2 карбоксильных групп, участвующих в реакциях (5) и (9), отвечает за увеличение длительности второго и третьего этапов взаимодействия. Максимальное содержание карбоксильных групп у образца 1 увеличивает скорость образования комплексов по (5) и адсорбции ГВ на поверхности наночастиц по (9). Следовательно, динамика взаимодействия ГВ с металлическим железом, помимо прочего, определяется их структурно-групповыми свойствами. Очевидно, что несмотря на многообразие реакций, сопровождающих взаимодействие ГВ и железа, варьируя структурно-групповые свойства ГВ, можно влиять на отдельные этапы этого взаимодействия. И наоборот, анализируя динамику изменения относительно легко контролируемых макропараметров взаимодействия ГВ и железа, можно сделать вывод о структурных свойствах природных ГВ.
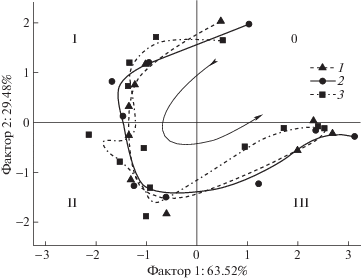
Выше было показано, что сложное многостадийное взаимодействие растворов ГВ и металлического железа может быть описано набором экспериментально определяемых параметров. Однако направления их изменений в ходе взаимодействия и их относительные связи не очевидны. Для редукции данных, описывающих развитие изучаемой системы, и для определения структуры взаимосвязей между переменными, использованных при построении рис. 4 г–е, был проведен анализ главных компонентов. Из результатов анализа (табл. 3) следует, что экспериментальные данные могут быть описаны с качеством представления в 93% с помощью двух основных компонентов (рис. 5). Причем проекции наблюдений на факторную плоскость для каждого эксперимента располагаются последовательно в форме С-образной кривой. Кроме того, наблюдения, отвечающие различным этапам взаимодействия железа с ГВ, располагаются строго в различных квадрантах факторной плоскости. Так, во втором координатном углу располагаются наблюдения, соответствующие первому этапу взаимодействия, в третьем – второму этапу, а в четвертом – третьему этапу (рис. 5). При этом наблюдения, соответствующие исходным растворам, группируются в первом координатном углу. Как следствие, можно указать на отрицательную корреляцию параметров, описывающих систему на первом и третьем этапах, а также параметров исходных растворов и второго этапа (рис. 5). Таким образом, обнаруживается однозначное соответствие между экспериментально определяемыми параметрами реакционной системы и предложенными этапами взаимодействия железа и ГВ. При этом предлагаемое преобразование экспериментальных данных позволяет непосредственно следить за “ходом реакций”, протекающих в изучаемой реакционной системе, и оценивать полноту превращения реагентов.
Таблица 3.
Факторные нагрузки, собственные значения и дисперсии главных компонент
| Компонента | Ф1 | Ф2 | Ф3 | Ф4 |
|---|---|---|---|---|
| Время, о.е. | 0.6480 | –0.7092 | 0.2717 | 0.0573 |
| (E254 – X*)/ϭ | –0.7622 | –0.5981 | –0.2013 | 0.1445 |
| (pH – X)/ϭ | –0.8635 | 0.4033 | 0.2778 | 0.1205 |
| (Eredox – X)/ϭ | 0.8913 | 0.3949 | –0.1005 | 0.1987 |
| Собственные значения | 2.5408 | 1.1794 | 0.2017 | 0.0782 |
| % от общей дисперсии | 63.52 | 29.48 | 5.04 | 1.95 |
| Кумулятивный % | 63.52 | 93.01 | 98.05 | 100.00 |
Рис. 5.
Проекции наблюдений на факторную плоскость Ф1*Ф2. Стрелкой указано направление развития изучаемых систем; I, II и III – первый, второй и третий этапы; 0 – исходные растворы.
Таким образом, на примере трех препаратов ГВ угля показано, что взаимодействия в системе “раствор ГВ – металлическое железо” осуществляются по единой схеме, включающей последовательность одинаковых реакций. Влиять на скорость протекания отдельных реакций можно, контролируя структурно-групповые свойства препаратов ГВ. Предложен математический аппарат контроля за ходом взаимодействия водных растворов ГВ с металлическим железом. Сделан вывод, что окислительный in situ синтез железосодержащих частиц [13] не зависит от метода производства и конкретного сырьевого источника и может быть масштабирован на препараты ГВ угля различных производителей.
Список литературы
Yan M., Sun C., XuJ. et al. // Corros. Sci. 2014. V. 80. P. 309.
Карпов В.А., Ковальчук Ю.Л., Харченко У.В., Беленева И.А. // Коррозия: материалы, защита. 2011. Т. 3. С. 11.
Колесникова Н.Н., Луканина Ю.К., Хватов А.В. и др. // Вестн. Казанск. технологич. ун-та. Химия. 2013. Т. 16. № 1. С. 170.
Sposito G. The Chemistry of Soils. New York: Oxford University Press, 2008. 342 p.
Santos A dos., Botero W.G., Bellin I.C. et al. // J. Braz. Chem. Soc. 2007. V. 18. № 4. P. 824.
Ghabbour E.A., Scheinost A.C., Davies G. // Chemosphere. 2007. V. 67. № 2. P. 285.
Gustafsson J.P. // J. Colloid Interface Sci. 2001. V. 244. № 1. P. 102.
Redman A.D., Macalady D., Ahmann D. // Environ. Sci. Technol. 2002. V. 36. № 13. P. 2889.
Zhou S., Chen S., Yuan Y., Lu Q. // Sci. Rep. 2015. V. 5. P. 1.
Amjad Z.// Int. J. Corros. Scale Inhib. 2017. V. 6. № 2. P. 162.
Vodyanitskii Y.N. // Eurasian Soil Sci. 2010. V. 43. № 11. P. 1244. Водяницкий Ю.Н. // Почвоведение. 2010. № 11. С. 1341.
Cole I.S., Marney D. // Corros. Sci. 2012. V. 56. P. 5.
Панкратов Д.А., Анучина М.М. // Журн. физ. химии. 2017. Т. 91. № 2. С. 234. Pankratov D.A., Anuchina M.M. // Russ. J. Phys. Chem. A. 2017. V. 91. № 2. P. 233.
Sorkina T.A., Polyakov A.Y., Kulikova N.A. et al. // J. Soils and Sediments. 2014. V. 14. № 2. P. 261.
Анучина М.М., Панкратов Д.А. // Агрохимический вестник. 2018. Спец. вып. С. 47.
Панкратов Д.А., Анучина М.М., Борисова Е.М. и др. // Журн. физ. химии. 2017. Т. 91. № 6. С. 1042. Pankratov D.A., Anuchina M.M., Borisova E.M. et al. // Russ. J. Phys. Chem. A. 2017. V. 91. № 6. P. 1109.
Kovalevskii D.V., Permin A.B., Perminova I.V., Petrosyan V.S. // Moscow University Chemistry Bulletin. 2000. V. 41. P. 39. Ковалевский Д.В., Пермин А.Б., Перминова И.В., Петросян В.С. // Вестн Моск. ун-та. Серия 2. Химия. 2000. Т. 41. С. 39.
Perminova I.V., Frimmel F.H., Kudryavtsev A.V., Kulikova N.A. // Environ. Sci. Technol. 2003. V. 37. № 11. P. 2477.
Соколов Р.С. Практические работы по химической технологии / Учебное пособие М.: Владос, 2004. 166 с.
Караванова Е.И. Оптические свойства почв и их природа. М.: Изд-во Моск. ун-та, 2003. 153 с.
Enev V., Pospisilova L., Klucakova M. et al. // Soil Water Res. 2014. V. 9. № 1. P. 9.
Weishaar J.L., Aiken G.R., Bergamaschi B.A. et al. // Environ. Sci. Technol. 2003. V. 37. P. 4702.
Uyguner C.S., Bekbolet M. // Catal. Today. 2005. V. 101. № 3–4. P. 267.
Mignone R.A., Martin M.V., Moran Vieyra F.E. et al. // Photochem. Photobiol. 2012. V. 88. № 4. P. 792.
Korshin G.V., Wu W.W., Benjamin M.M., Hemingway O. // Water Res. 2002. V. 36. P. 3273.
Pourbaix M. // Corros. Sci. 1990. V. 30. № 10. P. 963.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии