

Журнал физической химии, 2019, T. 93, № 8, стр. 1164-1168
Константы устойчивости глицилглицинатных комплексов меди(II) в водно-этанольных растворах
В. А. Исаева a, *, А. С. Молчанов b, К. А. Кипятков b, В. А. Шарнин a
a Ивановский государственный химико-технологический университет
Иваново, Россия
b Костромской государственный университет
Кострома, Россия
* E-mail: kvol1969@gmail.com
Поступила в редакцию 04.10.2018
После доработки 23.12.2018
Принята к публикации 23.12.2018
Аннотация
Потенциометрическим методом определены константы устойчивости глицилглицинатных комплексов меди(II) в водно-этанольных растворах переменного состава при температуре 298 K и ионной силе растворов 0.1 (NaClO4). Установлено, что в растворе возможно образование нормальных и депротонированных моно- и бисглицилглицинатных комплексов меди(II). С увеличением концентрации органического компонента в растворе устойчивость глицилглицинатов меди(II) возрастает. Рассчитаны константы депротонирования пептидной группы глицилглицина. Дана оценка сольватационных вкладов реагентов в изменение энергии Гиббса изучаемых равновесных процессов при изменении концентрации этанола в растворе.
Анион глицилглицина способен образовывать комплексы с ионами d-металлов благодаря наличию нескольких центров координации. Процесс комплексообразования глицилглицинат-иона с ионом Cu(II) имеет особенность, связанную с протеканием реакции депротонирования пептидной группы, что обуславливает возможность образования в системе как нормальных, так и депротонированных глицилглицинатов меди(II) [1, 2]. В водных растворах устойчивость глицилглицинатных комплексов меди(II) изучена достаточно хорошо [1–4 и др.], в водно-органических растворах устойчивость глицилглицинатов Cu(II) охарактеризована единичными данными для водно-диоксановых смесей [5, 6]. В настоящей работе поставлена задача исследовать влияние состава водно-этанольного растворителя на смещение равновесных процессов, протекающих при комплексообразовании Cu2+ с глицилглицинат-ионом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для определения констант равновесия реакций комплексообразования меди(II) с глицилглицинат-ионом методом потенциометрического титрования использовали стеклянный электрод, на работоспособность которого в водно-этанольных смесях указывается в работе [7]. В качестве электрода сравнения применялся хлорсеребряный электрод. Для уменьшения диффузионного потенциала на концах электролитического мостика внутренний раствор электрода сравнения готовился на основе водно-этанольного растворителя соответствующего состава. Измерения выполнялись при температуре 298 K и ионной силе µ = 0.1 M на фоне перхлората натрия.
В ячейку заливали водно-этанольный раствор, содержащий Cu(ClO4)2 (8 × 10–3 моль/л) и HClO4 (1 × 10–3 моль/л). В качестве титранта использовали раствор глицилглицината натрия (5 × 10–1 моль/л). Дозировку титранта осуществляли весовым способом с помощью микрошприца. Титрование проводили в диапазоне рН 3.8–8.3.
В работе использовали Cu(ClO4)2, полученный из основного карбоната меди(II) “х. ч.” и хлорной кислоты “х. ч.” с последующей перекристаллизацией. Концентрацию полученного раствора перхлората меди(II) определяли титрованием этилендиаминацетатом натрия (ЭДТА). Перхлорат натрия (“ч.”) очищали перекристаллизацией из водного раствора. Раствор глицилглицината натрия готовили по точным навескам эквимолярных количеств глицилглицина (фирмы “Sigma” с содержанием основного вещества ≥99%) и бескарбонатного насыщенного раствора NaOH. Гидроксид натрия имел квалификацию “х. ч.”. Этанол (EtOH) марки “ректификат” перегоняли, остаточное содержание воды учитывали при приготовлении растворов.
Расчет констант устойчивости комплексов по результатам потенциометрического титрования проводили по программе PHMETR [8]. Погрешность численных значений констант оценивали на основе статистической обработки результатов серии измерений.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Глицилглицин  представляет собой дипептид, построенный из двух молекул аминокислоты глицина посредством
пептидной связи и существующий в водном растворе в виде цвиттер-иона (HGG±). Анион глицилглицина (GG–) образует комплексы с ионом двухвалентной меди посредством азота аминогруппы и кислорода
пептидной группы [1, 2, 9]. При образовании депротонированных комплексов Cu(II) с глицилглицинат-ионом, связанном
с диссоциацией пептидной группы лиганда, координация смещается с пептидного кислорода
на пептидный азот [1, 2].
представляет собой дипептид, построенный из двух молекул аминокислоты глицина посредством
пептидной связи и существующий в водном растворе в виде цвиттер-иона (HGG±). Анион глицилглицина (GG–) образует комплексы с ионом двухвалентной меди посредством азота аминогруппы и кислорода
пептидной группы [1, 2, 9]. При образовании депротонированных комплексов Cu(II) с глицилглицинат-ионом, связанном
с диссоциацией пептидной группы лиганда, координация смещается с пептидного кислорода
на пептидный азот [1, 2].
При взаимодействии Cu2+ с глицилглицинат-ионом в диапазоне рН 2–14 возможно образование порядка 16-ти комплексных частиц [3, 9]. Однако данные ЭПР исследований [10] не подтвердили образование биядерных комплексов меди(II) с анионом глицилглицина. Образование некоторых моноядерных комплексов Cu2+ с глицилглицинат-ионом авторы [4] не считают строго доказанным в виду того, что они не накапливаются в растворе в заметных количествах. Согласно анализa литературных данных [3, 4] в системе Cu2+ – глицилглицинат-ион наиболее вероятно образование нормального моноглицилглицината меди(II) [CuGG]+, депротонированного моноглицилглицината меди(II) [CuGG-H] и бисглицилглицината меди(II) [CuGG-HGG]–, содержащего одну депротонированную группу. Не исключается возможность образования комплекса Cu2+ с недиссоциированным глицилглицином [CuНGG]2+ [4], нормального бисглицилглицината меди(II) [CuGG2] [3, 4]. При высоких значениях рН возможно накопление гидроксочастиц. Для уточнения модели равновесий в системе провели расчет равновесного состава смеси в водном растворе по программе RRSU [11], учитывая возможность протекания следующих реакций:
(1)
${\text{G}}{{{\text{G}}}^{--}} + {{{\text{H}}}^{ + }} \leftrightarrow {\text{HG}}{{{\text{G}}}^{ \pm }},\quad \lg {{K}_{1}},$(2)
${\text{G}}{{{\text{G}}}^{--}} + 2{{{\text{H}}}^{ + }} \leftrightarrow {{{\text{H}}}_{2}}{\text{G}}{{{\text{G}}}^{ + }},\quad \lg {{\beta }_{2}},$(3)
${\text{G}}{{{\text{G}}}^{--}} + {\text{C}}{{{\text{u}}}^{{2 + }}} \leftrightarrow {{[{\text{CuGG}}]}^{ + }},\quad \lg {{K}_{3}},$(4)
${\text{G}}{{{\text{G}}}^{--}} + {\text{C}}{{{\text{u}}}^{{2 + }}}{\kern 1pt} --{\kern 1pt} {{{\text{H}}}^{ + }} \leftrightarrow [{\text{CuG}}{{{\text{G}}}_{{--{\text{H}}}}}],\quad \lg {{\beta }_{4}},$(5)
$2{\text{G}}{{{\text{G}}}^{--}} + {\text{C}}{{{\text{u}}}^{{2 + }}}{\kern 1pt} --{\kern 1pt} {{{\text{H}}}^{ + }} \leftrightarrow {{[{\text{CuG}}{{{\text{G}}}_{{ - {\text{H}}}}}{\text{GG}}]}^{--}},\quad \lg {{\beta }_{5}},$(6)
${\text{2G}}{{{\text{G}}}^{--}} + {\text{C}}{{{\text{u}}}^{{2 + }}} \leftrightarrow [{\text{CuG}}{{{\text{G}}}_{2}}],\quad \lg {{\beta }_{6}},$(7)
${\text{G}}{{{\text{G}}}^{--}} + {\text{C}}{{{\text{u}}}^{{2 + }}} + {{{\text{H}}}^{ + }} \leftrightarrow {{[{\text{CuHGG}}]}^{{2 + }}},\quad \lg {{\beta }_{7}},$(8)
${\text{G}}{{{\text{G}}}^{--}} + {\text{C}}{{{\text{u}}}^{{2 + }}}{\kern 1pt} --{\kern 1pt} 2{{{\text{H}}}^{ + }} \leftrightarrow {{[{\text{CuG}}{{{\text{G}}}_{{--{\text{H}}}}}{\text{OH}}]}^{--}},\quad \lg {{\beta }_{8}},$(9)
${\text{C}}{{{\text{u}}}^{{2 + }}} + {\text{O}}{{{\text{H}}}^{--}} \leftrightarrow {{[{\text{CuOH}}]}^{ + }},\quad \lg {{K}_{9}},$(10)
${{{\text{H}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}} \leftrightarrow {{{\text{H}}}_{2}}{\text{O}},\quad \lg {{K}_{{10}}}.$В расчетах использовали термодинамические константы образования нормальных и депротонированных моно- и бисглицилглицинатов меди(II) [3], константы протонирования глицилглицинат-иона [12], автопротолиза воды [13], процессов образования [CuHGG]2+, [CuGG–HOH]–, [CuOH]+ из работ [14–16] соответственно. Результаты определения равновесного состава смеси по программе RRSU показали отсутствие либо пренебрежимо малое накопление в системе [CuHGG]2+ и гидроксочастиц, что позволило исключить процессы их образования из расчетной схемы.
Таким образом при определении констант комплексообразования меди(II) с глицилглицинат-ионом по данным потенциометрического титрования в водных и водно-этанольных растворах по программе PHMETR [8] учитывали образование нормальных и депротонированных моно- и бисглицилглицинатных комплексов меди(II). В расчетную схему также включали реакции кислотно-основных равновесий глицилглицина и автопротолиза растворителя, константы которых для водно-этанольных растворов взяты из работ [12, 13] соответственно. Проверочные расчеты по программе PHMETR [8] для водно-этанольных смесей показали, что включение в расчетную схему процессов образования [CuHGG]2+ либо гидроксокомплексов не влияет на величины рассчитываемых констант (lg K3, lg β4, lg β5, lg β6), а наличие глицилглицинового и гидроксокомплексов в равновесном составе смеси не подтверждается.
Для водного раствора полученные нами константы образования нормальных и депротонированных моно- и бисглицилглицинатных комплексов меди(II) хорошо согласуются с литературными значениями для аналогичных условий (µ = = 0.1 M, Т = 298 K) (табл. 1) и незначительно отличаются от отнесенных к стандартным условиям констант, рекомендованных в [3] на основе критического анализа литературных данных.
Таблица 1.
Константы образования глицилглицинатных комплексов меди(II) и депротонирования пептидной группы глицилглицината меди(II) (рКа) в водном растворе, Т = 298 K
| lg K3 [CuGG]+ |
lg β4 [CuGG–H] |
lg β5 [CuGG–HGG]– |
lg β6 [CuGG2] |
рKа | µ | Источник |
|---|---|---|---|---|---|---|
| 5.56 | 1.46 | 4.64 | 11.00 | 4.10 | 0.1 (NaClO4) | Наши данные |
| 5.68 | 1.47 | 4.31 | – | 4.21 | 0.1 (КNO3) | [14] |
| 5.56 | 1.50 | – | – | 4.06 | 0.1 (NaClO4) | [15] |
| 5.40 | 1.47 | 5.14 | – | 3.93 | 0.1 (КCl) | [17] |
| 5.44 | 1.25 | – | – | 4.19 | 0.1 (КCl) | [18] |
| 5.55 | 1.56 | – | – | 3.99 | 0.1 (NaClO4) | [19] |
| 5.71 | 1.56 | – | – | 4.15 | 0.1 (NaClO4) | [20] |
| 5.68 | 1.50 | – | – | 4.18 | 0.1 (КNO3) | [21] |
| 5.56 | 1.44 | 4.61 | – | 4.12 | 0.1 (КNO3) | [22] |
| 5.43 | 1.26 | – | – | 4.17 | 0.1 (NaCl) | [23] |
| 5.97 | 1.84 | 5.14 | 11.14 | 4.13 | 0.0 | [3] |
Константы устойчивости глицилглицинатных комплексов меди(II) в водно-этанольных растворах приведены в табл. 2. Полученные значения констант равновесия процессов комплексообразования позволили рассчитать константу депротонирования пептидной группы в глицилглицинате меди(II): lg Kа = –рKа = lg β4 – lg K3.
Таблица 2.
Константы образования глицилглицинатных комплексов меди(II) и депротонирования пептидной группы глицилглицината меди(II) (рKа) в водно-этанольных растворах, Т = 298 K, µ = 0.1(NaClO4)
| Константа равновесия | Комплексная частица | Состав растворителя, мол. доли этанола | ||||||
|---|---|---|---|---|---|---|---|---|
| 0.0 | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | ||
| lg K3 ± 0.05 | [CuGG]+ | 5.56 | 6.10 | 6.56 | 6.83 | 7.06 | 7.56 | 8.11 |
| lg β4 ± 0.08 | [CuGG–H] | 1.46 | 1.92 | 2.11 | 2.14 | 2.24 | 2.54 | 2.80 |
| lg β5 ± 0.08 | [CuGG-HGG]– | 4.64 | 5.45 | 6.05 | 6.21 | 6.61 | 7.28 | 8.23 |
| lg β6 ± 0.08 | [CuGG2] | 11.00 | 11.71 | 12.39 | 12.81 | 13.19 | 14.02 | 14.70 |
| рKa ± 0.08 | 4.10 | 4.18 | 4.45 | 4.69 | 4.82 | 5.02 | 5.31 | |
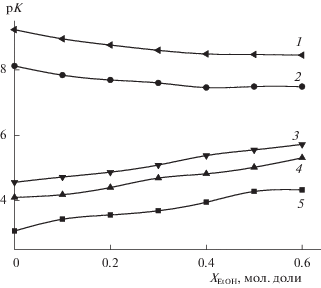
Для водного раствора величина рKа, определенная в настоящей работе, находится в соответствии с литературными значениями (табл. 1). Добавление в водный раствор этанола приводит к изменению рKа (табл. 2). Известно, что с ростом концентрации этанола в растворе рK диссоциации концевой протонированной аминогруппы цвиттер-иона глицилглицина уменьшается [12], как и рK протонированных аминов [24] (рис. 1). Депротонирование пептидной группы в глицилглицинате меди(II) идет по типу диссоциации незаряженных кислот и линейно возрастающий характер зависимости рKа = f(EtOH) пептидной группы соответствует аналогичным зависимостям для процессов диссоциации карбоксильной группы глицилглициний-иона [12], а также карбоновых кислот [25] (рис. 1).
Наблюдаемое возрастание констант устойчивости глицилглицинатов меди(II) в водно-этанольных растворах (табл. 2), характерно для большинства реакций образования комплексов d-металлов с N-, O-донорными лигандами [26–30 и др.]. Сопоставление устойчивости комплексов меди(II) с простейшими представителями карбоновых кислот, аминокислот и пептидов показывает (рис. 2), что как в водных, так и водно-этанольных растворах благодаря наличию нескольких центров координации глицинатные [27] и глицилглицинатные комплексы обладают большей устойчивостью, чем ацетатные [29]. В свою очередь устойчивость комплексов рассматриваемых лигандов с ионом меди(II) существенно превышает устойчивость этих же комплексов с ионом никеля(II) [12, 28, 30] (рис. 2). В глицилглицинатных комплексах усиление связи металл-лиганд способствует более легкому отщеплению пептидного водорода и приводит к тому, что диссоциация пептидной группы в глицилглицинате Cu2+ происходит в области рН 5–7 с рKа = 1.84 (µ = 0.0), в глицилглицинате Ni2+ при рН 10 с рKа = –3.02 (µ = 0.0) [3].

Увеличение содержания этанола в растворе наиболее существенно изменяет устойчивость моноглицилглицинатных комплексов (табл. 3). Из того, что водно-этанольный растворитель оказывает незначительное влияние на устойчивость аммиачных комплексов и заметно изменяет устойчивость ацетатных и глицинатных комплексов [26] следует, что упрочнение глицинатов меди(II) и никеля(II) в водно-этанольных растворах происходит за счет упрочнения связи ионов металлов с карбоксилатной группой. Поскольку в образовании нормальных моноглицилглицинатных комплексов участвует амино- и пептидная группы [1, 2, 9] и учитывая соотношение прироста констант устойчивости комплексов (табл. 3), можно полагать, что в водно-этанольных растворителях прочность связи иона металла с кислородом пептидной группы в комплексных частицах увеличивается в большей мере, чем с кислородом карбоксилатной группы.
Таблица 3.
Изменение констант устойчивости монолигандных комплексов никеля(II) и меди(II) в диапазоне составов растворителя 0.0–0.5 мол. долей этанола
Для выявления причин смещения равновесия реакций комплексообразования в водно-органических растворителях обычно рассматривают вклады изменения термодинамических параметров пересольватации участников равновесия в изменение энергии Гиббса реакции [26]. Однако формальное использование сольватационного подхода к рассмотрению причин смещения химического равновесия в смешанных растворителях может привести к неверным выводам. Как показывает рис. 3, при образовании нормального моноглицилглицинатного комплекса меди(II) в водно-этанольном растворе вклады от изменения сольватного состояния комплексной частицы и лиганда взаимокомпенсируют друг друга, а ослабление сольватации Cu2+ [31] способствует росту устойчивости [CuGG]+ (рис. 3). Однако в действительности определяющим фактором смещения равновесия процесса комплексообразования с глицилглицинат-ионом в смешанном растворителе изменение сольватного состояния иона-комплексообразователя не является, так как устойчивость глицилглицината никеля(II) в водно-этанольных растворах также возрастает [12] при том, что ∆Gο пересольватации Ni2+ характеризуется отрицательным значением при содержании этанола 0.15 мол. долей [32]. Согласно [26] определяющий вклад в упрочнение комплексных частиц вносит изменение сольватного состояния лиганда в водно-органических растворах. Известно, что ∆trG переноса из воды в водно-этанольный растворитель лигандов карбоксилатного типа характеризуется высоким положительным значением [33, 34]. Значительное ослабление сольватации карбоксилат-ионов в водно-органическом растворе определяет рост устойчивости их комплексов с ионами d-металлов.
Рис. 3.
Влияние состава водно-этанольного растворителя на: 1 – ∆trG°(Cu2+) [31], 2 – (∆trG([CuGG]+) – ∆trG(GG–)), 3 – ∆trG реакции образования [CuGG]+.

Список литературы
Datta S.P., Rabin B.R. // Trans. Faraday Society. 1956. V. 52. P. 1117.
Nakon R., Angelici R.J. // Inorg. Chem. 1973. V. 12. № 6. P. 1269.
Кочергина Л.А., Емельянов А.В. // Журн. физ. химии. 2015. Т. 89. № 4. С. 592.
Гоголашвили Э.Л., Захаров А.В., Фаррахова Г.А. // Изв. вузов. Химия и хим. технология. 1983. Т. 26. № 1. С. 10.
Pelletier S.J. // Chim. Phys. 1972. V. 69. P. 751.
Chakraborty D., Bhattacharya P. // J. Inorg. Biochem. 1991. V. 41. P. 57.
Бургер К. Сольватация, ионные реакции и комплексообразование в неводных средах. М.: Мир, 1984. 256 с.
Бородин В.А., Козловский Е.В., Васильев В.П. // Журн. неорган. химии. 1986. Т. 31. № 1. С. 10.
Эйхгорн Г. Неорганическая химия. Т. 1 / Пер. с англ. под ред. М.Е. Вольпина, К.Б. Яцимирского. М.: Мир, 1978. 713 с.
Lafferty F.L., Jensen E.G., Sheppard J.G. // Inorg. Ghem. 1969. V. 8. № 9. P. 1875.
Васильев В.П., Бородин В.А., Козловский Е.В. Применение ЭВМ в химико-аналитических расчетах. М.: Высш. школа, 1993. 112 с.
Наумов В.В., Исаева В.А., Шарнин В.А. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1208.
Woollej E.H., Hurkot D.G., Herber L.G. // J. Phys. Chem. 1970. V. 74. № 22. P. 3908.
Kaneda A., Martell A. // J. Coord. Chem. 1974. V. 4. P. 137.
Brunetti A., Lim M., Nancollas G. // J. Am. Chem. Soc. 1968. V. 90. P. 5120.
Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1971. 456 с.
Bordignon-Luiz M., Szpoganicz B., Rizzoto M. et al. // Inorg. Chim. Acta. 1997. V. 254. P. 345.
Яцимирский К.Б., Манорик П.А., Давиденко Н.К. // Координац. химия. 1988. Т. 14. № 3. С. 311.
Sigel H., Prijs B., Martin R. // Inorg. Chim. Acta. 1981. V. 56. P. 45.
Sigel H., Grisser R., Prijs B. // Z. Naturforsch. 1972. B. 27B. S. 353.
Yamauchi O., Hirano Y., Nakao Y., Nakahara A. // Can. J. Chem. 1969. V. 47. P. 3441.
Martin R. // Bull. Soc. Chim. Fr. 1967. P. 2217.
Biester J.L., Ruoff P.M. // J. Am. Chem. Soc. 1959. V. 81. P. 6517.
Афанасьев В.Н., Шорманов В.А., Крестов Г.А. // Тр. Ивановского хим.-технолог. ин-та. 1972. Вып. 13. С. 36.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Журн. физ. химии. 1997. Т. 71. № 8. С. 1371.
Шарнин В.А. // Изв. вузов. Химия и хим. технология. 2005. Т. 48. Вып. 7. С. 44.
Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Журн. неорган. химии. 1997. Т. 42. № 7. С. 1220.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Координац. химия. 1999. Т. 25. № 12. С. 912.
Sigel H., Malini-Balakrishava R., Hiring O.K. // J. Am. Chem. Soc. 1985. V. 107. № 18. P. 5137.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Журн. физ. химии. 1998. Т. 72. № 12. С. 2182.
Lewandowski A. // Electrochim. Acta. 1984. V. 29. P. 547.
Невский А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Координац. химия. 1983. Т. 9. № 3. С. 391.
Wells C.F. // J. Chem. Soc. Faradey Trans. 1979. V. 75. P. 53.
Dey B.P., Lahiri S.C. // Indian J. Chem. 1986. V. 25A. № 2. P. 136.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии