

Журнал физической химии, 2020, T. 94, № 1, стр. 16-20
Термодинамические характеристики пересольватации глицилглицинат-иона и их вклад в изменение устойчивости комплексов с Сu(II) и Ni(II) в водно-органических растворителях
В. А. Исаева a, *, А. С. Молчанов b, К. А. Кипятков b, В. А. Шарнин a
a Ивановский государственный химико-технологический университет
Иваново, Россия
b Костромской государственный университет
Кострома, Россия
* E-mail: kvol1969@gmail.com
Поступила в редакцию 18.04.2019
После доработки 05.07.2019
Принята к публикации 05.07.2019
Аннотация
Представлены определенные экспериментально и рассчитанные на основе собственных и литературных данных термодинамические характеристики (∆G, ∆Н, T∆S) пересольватации глицилглицина, его протонированной и депротонированной форм в водных растворах этанола и диметилсульфоксида. Рассмотрено изменение энергии Гиббса реакций образования глицилглицинатных комплексов никеля(II) и меди(II) в водно-этанольных и водно-диметилсульфоксидных растворителях и дана оценка сольватационных вкладов реагентов в изменение устойчивости комплексных частиц. Показаны особенности в изменениях термодинамических характеристик сольватации и комплексообразования глицилглицинат-иона с d-металлами в водно-органических смесях.
Большое количество реакций, осуществляемых в неводных и водно-органических растворах, относится к процессам комплексообразования нейтральных либо ионных форм лигандов с ионами металлов. Для выявления причин, вызывающих смещение равновесия процессов с изменением природы среды, универсальное значение имеет подход, который основан на рассмотрении термодинамических параметров пересольватации реагентов и продуктов межчастичных взаимодействий при варьировании состава растворителя [1]. Анализ результатов исследований процессов комплексообразования аминов, ацетат- и глицинат-ионов с ионами d-металлов в различных водно-органических растворителях показал [2], что упрочнение комплексов определяется, главным образом, ослаблением сольватации лиганда при частичной или полной компенсации сольватационных вкладов комплексного и центрального ионов.
В настоящей работе рассмотрено изменение сольватного состояния глицилглицинат-иона в водно-этанольных и водно-диметилсульфоксидных смесях и дана оценка вклада пересольватации лиганда в изменение устойчивости его комплексов с ионами меди(II) и никеля(II) при варьировании состава растворителя.
Изменение энтальпии пересольватации глицилглицинат-иона в водно-этанольных и водно-диметилсульфоксидных растворителях определено нами в работах [3, 4] калориметрическим методом, изменение энергии Гиббса переноса глицилглицина и его аниона из воды в смеси вода–диметилсульфоксид определено методом межфазного распределения [5].
Значения изменения энергии Гиббса переноса глицилглицина (HGG±) из воды в растворитель вода–этанол переменного состава были рассчитаны по уравнению:
(1)
${{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{HG}}{{{\text{G}}}^{ \pm }}) = --2.303RT\lg ({{C}_{{{\text{mix}}}}}{\text{/}}{{C}_{{\text{w}}}}),$Растворимость глицилглицина (мг/мл) при 298 K приведена в работе [6] в диапазоне составов водно-этанольного растворителя 0–60% (об.). Данные [6] пересчитаны в молярную концентрацию (моль/л) и приведены к шкале содержания этанола в растворе, выраженного в мольных долях через плотность водно-этанольных смесей при 298 K [7]. Используя полученные ΔtrG°(HGG±), значения изменения энергии Гиббса реакций диссоциации глицилглициний-иона (ΔtrGr1) и глицилглицина (ΔtrGr2), а также литературные данные о ΔtrG° протона в водно-этанольном растворителе [8] рассчитали значения ΔG° переноса из воды в водно-этанольный растворитель глицилглицинат-иона (GG–) и глицицилглициний-иона (Н2GG+):
(2)
$\begin{gathered} {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r}}1}}^{ \circ } = {{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{HG}}{{{\text{G}}}^{ \pm }}) + {{\Delta }_{{{\text{tr}}}}}G^\circ ({{{\text{H}}}^{ + }})-- \\ - \;{{\Delta }_{{{\text{tr}}}}}G^\circ ({{{\text{H}}}_{2}}{\text{G}}{{{\text{G}}}^{ + }}), \\ \end{gathered} $(3)
$\begin{gathered} {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2}}}}^{ \circ } = {{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{G}}{{{\text{G}}}^{ - }}) + {{\Delta }_{{{\text{tr}}}}}G^\circ ({{{\text{H}}}^{ + }})-- \\ - \;{{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{HG}}{{{\text{G}}}^{ \pm }}). \\ \end{gathered} $Значения изменений энергии Гиббса реакций образования глицилглицинатных комплексов никеля(II) и меди(II) в водно-этанольных и водно-диметилсульфоксидных растворителях рассчитаны по константам устойчивости глицилглицинатов Ni2+ и Cu2+, определенным потенциометрическим методом при Т = 298 K, µ = 0.1 (NaClO4) [9–11]. Расчет изменения энергии Гиббса переноса из воды водно-этанольный растворитель комплексных частиц [CuGG]+ и [NiGG]+ проводили по уравнению:
(4)
$\begin{gathered} {{\Delta }_{{{\text{tr}}}}}G_{{\text{r}}}^{ \circ } = {{\Delta }_{{{\text{tr}}}}}G^\circ ({{[{\text{МеGG}}]}^{ + }})--{{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{М}}{{{\text{е}}}^{{2 + }}}) - \\ --\;{{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{G}}{{{\text{G}}}^{ - }}), \\ \end{gathered} $Значения энергии Гиббса переноса из воды в водно-этанольные и водно-диметилсульфоксидные смеси глицилглицинат-иона, глицилглицина и глицилглициний-иона, рассчитанные по уравнениям (1)–(3) и представленные в табл. 1, показывают, что в смешанном растворителе происходит ослабление сольватации глицилглицина и его аниона. Являясь бидентатным лигандом, глицилглицинат-ион имеет в своем составе карбоксилатную и аминогруппы, каждая из которых оказывает влияние на величину ΔG° пересольватации аниона дипептида в водно-органическом растворителе. Можно полагать, что доминирующий вклад в величину энергии Гиббса переноса глицилглицинат-иона из воды в смеси вода–этанол и вода–диметилсульфоксид вносит карбоксилатная группа. Такое предположение основано на данных работ [14–16], в которых показано, что значение ΔG° переноса лигандов карбоксилатного типа из воды в смешанный растворитель существенно возрастает, тогда как для аминов (аммиака) в том же интервале составов водно-органического растворителя изменение энергии Гиббса пересольватации незначительно [17, 18] (рис. 1).
Таблица 1.
Термодинамические параметры пересольватации глицилглицина, глицилглициний-иона и глицилглицинат-иона в водно-этанольном и водно-диметилсульфоксидном растворителях при 298 K, кДж/моль
| Параметр | Концентрация органического компонента, мол. доли | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.8 | 0.99 | |
| вода–этанол | |||||||||
| ΔtrG°(HGG±) | 1.6 | 3.3 | 6.6 | 8.7 | |||||
| ΔtrG°(H2GG+) | 1.0 | 1.0 | 1.7 | 1.0 | |||||
| ΔtrG°(GG–) | 1.2 | 2.1 | 6.3 | 9.9 | |||||
| ΔtrH°(GG–) | 1.9 | 2.8 | 5.9 | 12.1 | 14.4 | 16.1 | 18.9 | 21.1 | |
| TΔtrS°(GG–) | 0.7 | 0.7 | –0.4 | 2.2 | |||||
| вода–диметилсульфоксид | |||||||||
| ΔtrG°(HGG±) | 1.15 | 3.04 | 3.36 | ||||||
| ΔtrG°(H2GG+) | –2.33 | –4.66 | –13.23 | ||||||
| ΔtrG°(GG–) | 2.32 | 5.66 | 11.32 | ||||||
| ΔtrH°(GG–) | 0.03 | 0.69 | 16.79 | 36.47 | 47.25 | 56.40 | 62.65 | 69.59 | 72.54 |
| TΔtrS°(GG–) | –2.3 | –5.0 | 5.5 | ||||||
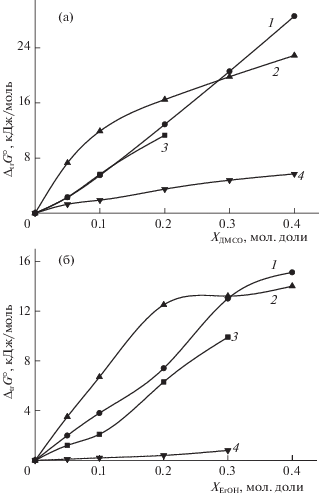
В работе [3] показано, что с ростом концентрации неводного компонента в растворе эндотермичность процесса пересольватации глицилглицинат-иона в водно-этанольном растворителе значительно возрастает, обусловливая ослабление сольватации аниона глицилглицина (табл. 1). Аналогичные изменения выявлены и для процесса пересольватации глицилглицинат-иона в растворителе вода–диметилсульфоксид [4] (табл. 1). Возрастающая в интервале более высоких концентраций как этанола, так и диметилсульфоксида, величина ΔtrH°(GG–) будет способствовать дальнейшему ослаблению сольватации лиганда, при этом роль энтропийной составляющей в изменении энергии Гиббса пересольватации глицилглицинат-иона в обоих смешанных растворителях в исследуемой области концентраций неводного компонента незначительна.
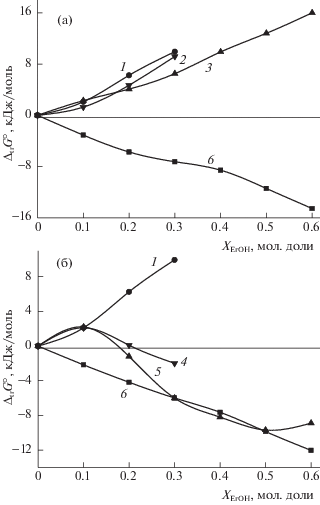
Глицилглицинат-ион образует с ионом никеля(II) комплексы состава 1 : 1, 1 : 2, 1 : 3. С ионом Cu2+ образуются моно- и бис-глицилглицинатные комплексы как нормальные, так и депротонированные, содержащие лиганд с диссоциированной в присутствии меди(II) пептидной группой [19, 20]. Исследования [9–11] показали, что в водных растворах этанола и диметилсульфоксида устойчивость глицилглицинатных комплексов никеля(II) и меди(II) при повышении концентрации неводного компонента в растворе возрастает. Причины повышения устойчивости комплексов с позиций сольватационного подхода рассмотрим на примере моноглицилглицинатов [CuGG]+ и [NiGG]+ в водно-этанольном растворителе. Как показывает рис. 2, изменение сольватного состояния иона-комплексообразователя в водно-этанольных смесях не является определяющим фактором упрочнения комплексных частиц. Изменение энергии Гиббса пересольватации Cu2+ [12] и Ni2+ [13] в водно-этанольных растворах имеет противоположную направленность при том, что изменения ΔG реакций образования [CuGG]+ и [NiGG]+ соизмеримы. Определяющий вклад в изменение устойчивости комплексов вносит ослабление сольватации глицилглицинат-иона. В работе [2] показано, что устойчивость комплексов d-металлов с аминами и карбоксилат-ионами в водно-органических растворителях не только зависит от сольватации лиганда, но и диапазон значений ${{\Delta }_{{{\text{tr}}}}}G_{{\text{r}}}^{ \circ }$ находится в пределах изменения функции лиганда (${{\Delta }_{{{\text{tr}}}}}G_{{\text{L}}}^{ \circ }$). Полученные зависимости для глицилглицинатных комплексов Cu2+ и Ni2+ (рис. 2) соответствуют этой общей закономерности.
Рис. 2.
Влияние водно-этанольного растворителя на изменение энергии Гиббса сольватации реагентов и реакций образования глицилглицинатов меди(II) (а) и никеля(II) (б); 1 – ΔtrG°(GG–), 2 – ΔtrG°([CuGG]+), 3 – ΔtrG°(Cu+) [12], 4 – ΔtrG°([NiGG]+), 5 – ΔtrG°(Ni2+) [13], 6 – ${{\Delta }_{{{\text{tr}}}}}G_{{\text{r}}}^{ \circ }$.
Соотношение термодинамических характеристик реакции комплексообразования ($\Delta Y_{{\text{r}}}^{ \circ }$ = = ($\Delta G_{{\text{r}}}^{ \circ }$, $\Delta H_{{\text{r}}}^{ \circ }$)) и изменения термодинамических свойств лиганда ($\Delta Y_{{\text{L}}}^{ \circ }$), как показано в работе [2], определяется коэффициентом различий (αdif):
(5)
$\Delta Y_{{\text{r}}}^{ \circ } = ({{\alpha }_{{{\text{dif}}}}}--1)\Delta Y_{{\text{L}}}^{ \circ }.$Таблица 2.
Значения коэффициентов различий для реакций образования комплексов меди(II) и никеля(II) с N- и О-донорными лигандами в водно-этанольных и водно-диметилсульфоксидных растворителях (х2 – концентрация органического компонента Х)
| Реакция | Х | х2, мол. доли | αdif |
|---|---|---|---|
| Cu2+ + GG– ↔ [CuGG]+ | EtOH | 0.3 | 0.3 |
| Ni2+ + GG– ↔ [NiGG]+ | EtOH | 0.3 | 0.4 |
| DMSO | 0.2 | 0.5 | |
| Cu2+ + NH3 ↔ [CuNH3]2+ | EtOH | 0.7 | 0.2 |
| Ni2+ + NH3 ↔ [NiNH3]2+ | EtOH | 0.6 | 0.5 |
| DMSO | 0.4 | 0.6 | |
| Cu2+ + Ac– ↔ [CuAc]+ | EtOH | 0.4 | 0.5 |
| Ni2+ + Ac– ↔ [NiAc]+ | EtOH | 0.4 | 0.7 |
| DMSO | 0.4 | 0.8 | |
| Cu2+ + Gly– ↔ [CuGly]+ | EtOH | 0.4 | 0.7 |
| Ni2+ + Gly– ↔ [NiGly]+ | EtOH | 0.4 | 0.7 |
| DMSO | 0.4 | 0.8 |
При образовании глицилглицинатных комплексов Ni(II) в растворителе вода–диметилсульфоксид значение αdif, рассчитанное по изменению энергии Гиббса реакции [10] и ∆Gο пересольватации лиганда [5] составило 0.5 (табл. 2). Коэффициент различий, рассчитанный через изменение энтальпии реакции образования глицилглицината Ni(II) [28] и изменение энтальпии пересольватации аниона глицилглицина [4] в растворителе вода–диметилсульфоксид, также имел значение 0.5. Ниже ожидаемого для карбоксилатных комплексов значения составили и величины αdif, определенные для процессов комплексообразования ионов металлов с глицилглицинат-ионом в растворителе вода–этанол по изменению энергии Гиббса реакций и ∆Gο пересольватации лиганда (табл. 2).
Таким образом, оценка вклада термодинамических параметров пересольватации глицилглицинат-иона в изменение устойчивости его комплексов с ионами меди(II) и никеля(II) в водно-этанольных и водно-диметилсульфоксидных растворителях показала некоторые отличия от процессов комплексообразования с другими лигандами карбоксилатного типа. При этом общие закономерности изменения термодинамических параметров реакций комплексообразования и пересольватации реагентов в водно-органических растворителях, установленные для аминных и карбоксилатных комплексов d-металлов нашли подтверждение для процессов образования глицилглицинатов меди(II) и никеля(II).
Список литературы
Комплексообразование в неводных средах / Г.А. Крестов, В.Н. Афанасьев, А.В. Агафонов и др. М.: Наука, 1989. 256 с.
Шарнин В.А. // Изв. вузов. Химия и хим. технология. 2005. Т. 48. Вып. 7. С. 44.
Исаева В.А., Наумов В.В., Шарнин В.А. // Журн. физ. химии. 2016. Т. 90. № 5. С. 721.
Наумов В.В., Исаева В.А., Шарнин В.А. // Там же. 2014. Т. 88. № 3. С. 443.
Наумов В.В., Исаева В.А., Кузина Е.Н., Шарнин В.А. // Там же. 2012. Т. 86. № 12. С. 1907.
Lu J., Wang X.-J., Yang X., Ching C.-B. // J. Chem. Eng. Data. 2006. V. 51. № 5. P. 1593.
Yilmaz H. // Turk. J. Phys. 2002. V. 26. P. 243.
Kalidas C., Hefter G., Marcus Y. // Chem. Rev. 2000. V. 100. № 3. P. 819.
Наумов В.В., Исаева В.А., Шарнин В.А. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1208.
Наумов В.В., Исаева В.А., Ковалева Ю.А., Шарнин В.А. // Журн. физ. химии. 2013. Т. 87. № 7. С. 1160.
Кипятков К.А., Молчанов А.С., Исаева В.А. // Сборник статей XVIII Междунар. научно-практич. конф. “Российская наука в современном мире”. 15 ноября 2018, Москва. М.: Научн.-издат. центр “Актуальность. РФ”, 2018. С. 46–47.
Lewandowski A. // Electrochim. Acta. 1984. V. 29. P. 547.
Невский А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Коорд. химия. 1983. Т. 9. № 3. С. 391.
Dey B.P., Lahiri S.C. // Indian J. Chem. A. 1986. V. 25. № 2. P. 136.
Wells C.F. // J. Chem. Soc. Farad. Trans. 1979. V. 75. P. 53.
Гессе Ж.Ф., Исаева В.А., Шарнин В.А. // Журн. физ. химии. 2010. Т. 84. № 2. С. 385.
Невский А.В., Шорманов В.А., Крестов Г.А. // Коорд. химия. 1983. Т. 9. № 3. С. 391.
Нищенков А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Журн. физ. химии. 1988. Т. 62. № 9. С. 2568.
Гоголашвили Э.Л., Захаров А.В., Фаррахова Г.А. // Изв. вузов. Химия и хим. технология. 1983. Т. 26. № 1. С. 10.
Кочергина Л.А., Емельянов А.В. // Журн. физ. химии. 2015. Т. 89. № 4. С. 592.
Нищенков А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Коорд. химия. 1990. Т. 16. № 9. С. 1264.
Исаева В.А., Шарнин В.А., Шорманов В.А., Леденков С.Ф. // Журн. физ. химии. 1996. Т. 70. № 7. С. 1320.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Там же. 1998. Т. 72. № 12. С. 2182.
Sigel H., Malini-Balakrishava R., Hiring O.K. // J. Am. Chem. Soc. 1985. V. 107. № 18. P. 5137.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Коорд. химия. 1999. Т. 25. № 12. С. 912.
Исаева В.А., Леденков С.Ф., Шарнин В.А., Шорманов В.А. // Журн. неорган. химии. 1994. Т. 39. № 12. С. 2028.
Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Там же. 1997. Т. 42. № 7. С. 1220.
Наумов В.В., Ковалева Ю.А., Исаева В.А., Усачева Т.Р., Шарнин В.А. // Журн. физ. химии. 2014. Т. 88. № 6. С. 969.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии