

Журнал физической химии, 2020, T. 94, № 1, стр. 98-107
Взаимодействие мезо-фенилзамещенных порфириноидов с карбоновыми кислотами и термический анализ их катионных солей
А. Е. Лихонина a, М. А. Крестьянинов b, Ф. К. Моршнев a, Е. Л. Критский a, Т. В. Кудаярова a, Д. Б. Березин a
a Ивановский государственный химико-технологический университет,
НИИ макрогетероциклических соединений
Иваново, Россия
b Российская академия наук, Институт химии растворов им. Г.А. Крестова
Иваново, Россия
Поступила в редакцию 13.03.2019
После доработки 16.03.2019
Принята к публикации 18.06.2019
Аннотация
Методами термогравиметрии и квантовой химии, включая NBO-анализ, изучены термоустойчивость и NH-кислотность одно- и двукратнопротонированных форм тетрапиррольных макрогетероциклических лигандов – порфиринов, их инвертированных и N-замещенных аналогов, а также корролов. Рассчитаны температурные интервалы деструкции ацетатов и трифторацетатов порфириния, энтальпии испарения молекул кислот из кристаллической фазы и состав катионных солей. Оптимизированы структуры протонированных форм макроциклов, рассчитаны энергии взаимодействия порфирин–кислота и величины переноса заряда при образовании связей.
Интерес к химии порфириноидов (НnР) – структурных аналогов порфиринов (Н2Р) – интенсивно растет в течение последних двух десятилетий. Эти соединения находят самое широкое применение в качестве перспективных составляющих новых материалов для фотоэлектроники, катализа, медицины и т.д. [1–6]. Вместе с тем, до настоящего времени неизвестны или не подвергнуты анализу некоторые их фундаментальные свойства, знание которых необходимо для успешного использования НnР.
Катионные формы порфиринов и их аналогов, принимающие участие во всех рН-зависимых физико-химических процессах, интенсивно исследуются в течение ряда десятилетий [7–10]. Наиболее востребованными методами исследования кислотно-основных равновесий являются спектрофотометрический и спектропотенциометрический [7, 11–14], а также фотофизические методы [10, 15]; в меньшей степени оказались задействованы калориметрический [16, 17] и квантово-химические [8, 9, 18, 19] методы. При этом в большинстве работ, посвященных исследованию кислотно-основных взаимодействий (КОВ) Н2Р и их аналогов, рассматривается состояние протонированных форм этих макрогетероциклов (МГЦ) в растворах. Свойства твердофазных катионных солей порфириния в политермических условиях ранее не изучались, обсуждались лишь процессы терморазложения кислотно-основных ассоциатов NH-активных Н2Р с электронодонорными молекулами [20].
Формально порфириноиды различных классов можно получить из собственно порфиринов, например, соединение I, путем проведения над их молекулами ряда операций, таких как N-замещение (II), сжатие полости МГЦ (III), изомеризация путем инверсии пиррольных циклов (IVab) или посредством изменения порядка чередования мезо-углеродных мостиков [2, 21].

Целью настоящей работы является изучение термоустойчивости катионных солей HnP, а также квантово-химический анализ глубины КОВ между макроциклами с различной конфигурацией, то есть числом и расположением кислотно-основных центров (I–IV), и молекулами карбоновых кислот (АсОН, TFA).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объекты исследования (I–IV) синтезированы, очищены и спектрально идентифицированы в соответствии с методиками, приведенными в литературе [22–25]. Трифторуксусную кислоту марки “х. ч.” (“Acros”) перегоняли с дефлегматором. Уксусную кислоту “х. ч.” (“Химреактив”) очищали согласно рекомендациям, описанным в [26]. Содержание воды в растворителях, оцененное титрованием по Фишеру, составило менее 0.1%.
Термогравиметрические исследования проводились на дериватографе, обеспечивающем синхронный термический анализ STA 449 F3 JUPITER (NETZSCH). Навеска кристаллического образца массой около 5 мг помещалась в платиновый тигель и нагревалась в статической атмосфере аргона со скоростью 5 K/мин в интервале температур 298–1223 K.
Образцы твердых катионных солей состава Н(n + 1)Р+Х– или Н(n + 2)Р2+(Х–)2, образующихся по уравнениям
(1)
${{{\text{H}}}_{n}}{\text{P}} + {\text{HX}} \rightleftharpoons {{{\text{Н}}}_{(}}{{_{n}}_{{ + 1)}}}{{{\text{Р}}}^{ + }}{{{\text{Х}}}^{--}},$(2)
${{{\text{H}}}_{n}}{\text{P}} + 2{\text{HX}} \rightleftharpoons {{{\text{Н}}}_{(}}{{_{n}}_{{ + 2)}}}{{{\text{Р}}}^{{2 + }}}{{({{{\text{Х}}}^{--}})}_{2}}$(3)
$\ln \left( {\frac{{dw}}{{d\tau }}{{T}^{{1/2}}}} \right) = - \frac{{{{\Delta }_{{{\text{vap}}}}}H}}{R}\frac{{{{{10}}^{3}}}}{T} + C,$Термогравиметрические опыты повторяли несколько раз, погрешность измерения температур этапов убыли массы (t, °С), а также расчета энтальпий испарения (ΔvapH) составляла не более 10%. Состав катионной соли рассчитывали по термокривой, исходя из массы навески, числа этапов терморазложения и убыли массы на каждом из них. Экспериментальные и расчетные данные приводятся в табл. 1.
Таблица 1.
Термодеструкция и состав катионных солей макрогетероциклов I–IV в инертной среде
| Соединение | Деструкция катионной соли порфириния | Мольное отношение ${{n}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$/nНХ/nМГЦ | Деструк-ция МГЦ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Испарение Н2О | Испарение НХ | Другие продукты разложения | |||||||||||||
| tн1, °C | tmax1, °C | tк1, °C | ∆vapH(1) кДж/моль | tн2, °C | tmax2, °C | tк2, °C | ∆vapH(2), кДж/моль | tн3, °C | tmax3, °C | tк3, °C | ∆vapH(3), кДж/моль | tн, °C | tпл, °C | ||
| АсOH | |||||||||||||||
| II | 79 | 97 | 116 | 33 | 128 | 142 | 156 | 53 | 198 | 211 | 224 | 203 | 4/2/1/1* | 427 | 421 |
| III | 69 | 87 | 108 | 29 | 130 | 149 | 162 | 46 | – | – | – | – | 6/1/1 | 308 | – |
| IV | 63 | 72 | 88 | 187 | – | – | – | – | – | – | – | – | 4/1** | 357 | 358 |
| TFA | |||||||||||||||
| I | 88 | 107 | 122 | 135 | 184 | 206 | 220 | 60 | – | – | – | – | 15/2/1 | 470 | – |
| II | 84 | 103 | 110 | 35 | 168 | 189 | 198 | 35 | – | – | – | – | 13/2/1 | 426 | – |
| III | 71 | 98 | 110 | 38 | 133 | 156 | 176 | 48 | – | – | – | – | 6/1/1 | 309 | – |
| IV | 72 | 103 | 114 | 47 | 185 | 204 | 216 | 99 | – | – | – | – | 7/1/1 | 354 | – |
Оптимизацию геометрических параметров макроциклов I–IV и продуктов их взаимодействия с одной или двумя молекулами уксусной и трифторуксусной кислот выполняли с использованием пакета программ Gaussian 09 [28], метода функционала плотности, гибридного функционала B3LYP [29] и базисного набора CC-pVDZ [30]. В рамках NBO-анализа были рассчитаны энергии взаимодействия кислотно-основных центров (Еint), а также энергии стабилизации орбитали протона, образующей водородную связь с донором электронной пары (Естаб), величины перенесенного на нее заряда (qстаб), геометрия реакционных центров Н-связанных форм МГЦ [31, 32]. Полное описание методики расчета приведено в работе [18], данные представлены в табл. 2.
Таблица 2.
Геометрические и энергетические параметры взаимодействия порфириноидов I–IV с карбоновыми кислотами
| Параметр | I · CH3COOH (2CH3COOH) |
I · CF3COOH (2CF3COOH) |
IVa · · CH3COOH (2CH3COOH) |
IVa · · CF3COOH (2CF3COOH) |
IVb · · CH3COOH (2CH3COOH) |
IVb · · CF3COOH (2 CF3COOH) |
II · · CH3COOH (2CH3 COOH) |
II · · CF3COOH (2CF3COOH) |
III · CH3 COOH | III· CF3 COOH | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Двугр угол Ψβ–β, град. | 9.9–16.3 (7.3–8.1) |
10.5–17.7 (8.3–9.4) |
14.5–22.1 (22.7–27.5) |
14.5–22.6 (25.2–28.5) |
8.9–19.4 (2.75–12.8) |
9.5–21.3 (0.66–12.4) |
15.8–39.0 | 15.8–38.7 (6.3–37.3) |
7.8–24.4 | 9.4–25.0 | |
| Двугр угол ΨN–N, град. | 0.937 (0) | 1.237 (0.002) | 4.8 (4.7) | 4.8 (4.9) | 6.4 (3.5) | 6.9 (4.3) | 4.7 | 4.8 (3.1) | 7.0 | 7.1 | |
| φPh, град. | 63.8–64.6 (67.4–67.9) |
63.2–69.3 (67.3–71.7) |
51.8–66.9 (48.6–63.4) |
51.7–67.2 (49.4–61.8) |
54.4–69.5 (59.3–81.9) |
52.8–69.6 (54.7–76.4) |
49.3–64.3 | 52.1–60.8 (53.1–79.0) |
47.0–60.5 | 47.2–59.3 | |
| –Е | 2142.99 (2372.10) |
2440.71 (2967.55) |
2142.97 (2372.05) |
2440.70 (2967.53) |
2142.88 (2372.06) |
2440.69 (2967.51) |
2182.17 | 2479.99 (3006.84) |
1873.80 | 2171.55 | |
| μ, D | 1.926 (0.002) |
4.106 (0.002) |
3.914 (4.809) |
8.440 (7.742) |
4.668 (2.083) |
3.207 (2.569) |
4.278 | 3.857 (2.026) |
1.348 | 2.266 | |
| ΔЕint | –26.69 (–48.28) |
–48.28 (–85.73) |
–46.94 (–73.26) |
–74.31 (–117.28) |
–35.27 (–58.45) |
–62.63 (–106.69) |
–34.18 | –59.25 (–80.67) |
–54.27 | –70.84 | |
| BSSE | 20.80 (40.74) |
19.95 (39.10) |
19.66 (40.18) |
18.91 (37.85) |
20.59 (41.06) |
19.59 (44.45) |
22.46 | 20.43 (38.41) |
23.76 | 22.04 | |
| $\Delta E_{{{\text{стаб}}}}^{{({\text{NBO}})}}$ | LP N → LP H–O LP N → BD H–O LP O → BD H–N BD N → BD H–N LP N → BD H–N |
73.05 (63.34/63.43) – – – – – – |
148.49 – (91.84/91.84) – – – – – |
190.25 (194.14/52.84) – – – – – – |
307.69 (303.51/70.17) – – – – – – |
69.58 (47.40/66.40) – – – – – – |
207.07 (153.47/220.58) – – – – – – |
108.95 – – – – – – – |
204.35 – (208.78/13.22) – – – – – |
– – 46.69 92.63 8.16 |
– – 120.37 77.86 8.07 |
| $q_{{{\text{стаб}}}}^{{({\text{NBO}})}}$ , ед.з. | LP N → LP H–O LP N → BD H–O LP N → BD N–H BD N → BD H–N LP N → BD H–N |
0.069 (0.059/0.059) – – – – – – |
0.0141 – (0.047/0.047) – – – – – |
0.171 (0.173/0.050) – – – – – – |
0.272 (0.269/0.037) – – – – – – |
0.035 (0.024/0.033) – – – – – –– |
0.192 (0.143/0.203) – – – – – – |
0.098 – – – – – – – |
0.185 – (0.190) (0.007) – – – – |
– – 0.221 0.042 0.072 |
– – 0.113 0.035 0.072 |
| φX ↔ Y–H, град. | N → N–H O → N–H N → H–O |
– – – – – 178.1 (173.6/173.6) |
– – – – – 177.5 (175.7/175.7) |
– – – – – 177.8 (177.9/162.7) |
– – – – – 177.2 (177.2/163.3) |
– – – – – 170.2 (172.2/172.1) |
– – – – – 172.0 (176.1/173.7) |
– – – – – 177.0 |
– – – – – 175.9 (177.1/162.4) |
136.4
– 163.8 166.8 |
135.5
– 163.8 168.0 |
| rX↔H, Å | N → N–H O → N–H N → H–O |
– – – – – 1.836 (1.857/1.857) |
– – – – – 1.683 (1.703/1.704) |
– – – – – 1.727 (1.722/1.857) |
– – – – – 1.597 (1.601/1.728) |
– – – – – 1.801 (1.853/1.800) |
– – – – – 1.635 (1.670/1.633) |
– – – – – 1.763 |
– – – – – 1.622 (1.626/1.979) |
1.804 – 1.810 1.794 |
1.822 – 1.855 1.651 |
| rX ↔ Y–H, Å | N → N–H O → N–H N → H–O |
– – – – – 2.833 (2.850/2.850) |
– – – – – 2.704 (2.721/2.721) |
– – – – – 2.739 (2.734/2.826) |
– – – – – 2.638 (2.640/2.717) |
– – – – – 2.795 (2.846/2.797) |
– – – – – 2.663 (2.694/2.664) |
– – – – – 2.768 |
– – – – – 2.655 (2.658/2.943) |
2.643 – 2.815 2.783 |
2.651 – 2.857 2.670 |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Конфигурация N-основных центров в молекулах порфиринов (соединение I) и их рассматриваемых в настоящей работе структурных аналогов (II–IV) существенно различается. Мезо-тетрафенилпорфин (I), как и большинство собственно Н2Р, в кислых средах образует симметричные N,N'-дикатионы (1), (2) [7–9, 11, 15, 16]. В молекулу N-замещенного порфириноида (II) вносит асимметрию внутрициклическая метильная группа, в результате чего N-основные центры становятся неэквивалентными [7, 12, 33–35]. Коррол (III) образует устойчивый N-монокатион, тогда как дальнейшее его протонирование по Смезо-углеродному атому с разрушением ароматической π-системы макроцикла возможно лишь в средах с высокой кислотностью (H2SO4, ТFA) [21]. Инвертированный порфириноид (IV) может существовать в виде двух таутомеров, в одном из которых оба N-основные центра являются внутрициклическими (IVb), а в другом один из них мигрирует на периферию молекулы (IVa) [12, 19, 33, 34].
Вопреки нашим ожиданиям неоднотипность и асимметричность локализации основных центров в молекулах порфириноидов II–IV не проявляется в увеличении числа пиков на кривых термодеструкции их катионных солей (1), (2). Обычно термограмма катионной соли порфириноида НnP (II–IV), аналогично термокривой порфириниевой соли (I, рис. 1, 2), включает три этапа деструкции: два в низкотемпературной области соответствуют отщеплению молекул связанной воды и кислоты, а последний, в области высоких температур, принадлежит терморазложению макрогетероцикла (рис. 1, табл. 1). Связанная вода, всегда присутствующая в составе катионной соли, в зависимости от природы МГЦ, начинает удаляться из ацетата порфириния при tн = 63–79°С с энтальпией испарения ΔvapH(1) = 29–33 кДж/моль, а из трифторацетата – при tн = 71–88°С с величиной ΔvapH(1) = 35–135 кДж/моль. При терморазложении ацетатов порфириния обычно выделяется в среднем от 2 до 6 молекул воды, тогда как в ходе термодеструкции их трифторацетатов – от 6 до 15 молекул. В расчете на одну молекулу воды средняя энтальпия ее испарения для различных НnP варьирует от 3 до 9 кДж/моль (табл. 1).

Рис. 2.
Термогравиметрические интегральная и дифференциальная кривые наноструктурированной дикатионной соли соединения IV состава (${{n}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ + nНХ)/nМГЦ = 4/1.

Испарение молекул кислот из катионных солей МГЦ I–IV начинается в интервале температур от 128 до 185°С, причем, в отличие от трифторуксусной, удаление уксусной кислоты практически не зависит от природы макроциклического основания (128–130°С; ΔvapH(2) = 46–53 кДж/моль, табл. 1).
Энтальпия испарения кислоты из соли порфириния в значительной мере определяется энергетикой взаимодействия третичных атомов азота МГЦ с протонами кислот, поэтому, с учетом числа испаряемых молекул АсОН, коррол III может рассматриваться как более основный МГЦ по сравнению с II (табл. 1).
В твердой фазе, в отличие от растворов, возможен также вклад в величину ΔvapH(2) эффектов кристаллической решетки. Так, ряд изменения энтальпии испарения TFA (ΔvapH(2), кДж/моль) в расчете на одну молекулу кислоты: IV (99) > III (48) > I (30) > II (17.5) согласуется с изменением основности макроциклов не в полной мере. Общеизвестным фактом является наименьшее сродство порфирина I к протону по сравнению с рассматриваемыми порфириноидами II–IV [21, 35, 36]. По-видимому, испарение связанной кислоты из кристалла дикатионной соли макроцикла II, имеющего сильно неплоскую структуру [33], существенно облегчается по сравнению с более плоским соединением I.
При этом в случае трифторуксусной кислоты число испаряемых из солей порфириния молекул НХ различно для разных МГЦ и равно двум в случае соед. I и II и единице для соед. III и IV (табл. 1). Действительно, порфирины и их N-замещенные аналоги являются двухкислотными основаниями, тогда как корролы в составе макроцикла имеют только один N-основный центр [21]. Полученные данные свидетельствуют о том, что, несмотря на образование корролом III дикатионной формы соли с участием третичного азота и мезо-углеродного атома в растворе концентрированной трифторуксусной кислоты [21], в кристаллическом состоянии она не существует, разрушаясь до N-монокатиона.
Неожиданным является тот факт, что инвертированный порфириноид IV, двухкислотное N‑основание, находящееся в условиях эксперимента в виде таутомера а [34], в ходе терморазложения его ацетатов и трифторацетатов отщепляет (т.е. изначально содержит) только одну молекулу кислоты.
Еще одной аномалией поведения IV в виде диацетата (табл. 1) является присутствие на его термокривой лишь двух этапов убыли массы (рис. 2). Высокотемпературный этап соответствует терморазложению МГЦ и вопросов не вызывает, тогда как первый наблюдается при температурах, близких к температурам испарения связанной воды (63°С), однако, величина энтальпии процесса при этом многократно превышает эту величину не только для воды, но и для кислот (187 кДж/моль). В случае трифторацетата IV ничего подобного не наблюдается. По-видимому, образование ацетатов IV благоприятствует формированию супрамолекулярной структуры на их основе, подобной описанной в работе [37]. Об этом же свидетельствуют существенные различия в электронных спектрах поглощения растворов IV в АсОН и TFA (рис. 3).

Третья стадия термодеструкции катионных солей порфириноидов связана с разложением макроциклических молекул I–IV в инертной атмосфере, которое начинается в температурном интервале 310–470°С. Например, в случае ацетатов I и IV оно происходит в области точки начала плавления твердого вещества, регистрируемой на кривой ДСК при 421 и 358°С (табл. 1, рис. 1) соответственно в виде острого эндо-пика, не сопровождающегося убылью массы образца.
Неизменность макроцикла после разложения катионной соли подтверждается хорошим соответствием температуры начала его разложения в составе соли (tн3, табл. 1) с tн индивидуального соединения [20, 21, 38]. Только в одном из изученных случаев (II) наблюдается частичное разложение МГЦ в ходе деструкции его диацетата. Разложение объясняется деалкилированием N-замещенного порфириноида II, на термокривой которого фиксируется еще одна стадия убыли массы с начальной температурой 198°С, согласующейся с литературными данными [38]. В случае трифторацетата II стадия деалкилирования отсутствует (табл. 1).
Согласно данным квантово-химических расчетов (DFT, B3LYP, базис CC-pVDZ), проведенных для идеализированных условий, взаимодействие лиганда НnP с одной или двумя молекулами кислоты ограничивается образованием Н-связей с высокой степенью переноса заряда (до 0.27 ед., табл. 2). Ранее [18, 19] расчеты в рассматриваемом базисе показали хорошее согласие с экспериментом для кислотно-основных форм NH-активных макроциклов с молекулами электронодоноров.
Оптимизация геометрии макроциклов I–IV с одной и (или) двумя молекулами АсОН и TFA, а также их NBO-анализ [31] позволили определить основные геометрические характеристики продуктов кислотно-основного взаимодействия, а также энергетические параметры этих взаимодействий для структур сходного строения. Так, согласно величине рассчитанной энергии взаимодействия МГЦ и НХ (Eint, кДж/моль, табл. 2) устойчивость моно- и дикатионов соед. I–IV с TFA возрастает в рядах (4) и (5), соответственно:
Приведенные ряды находятся в согласии с рядом основности НnР, полученным термогравиметрическим методом (см. выше). Вместе с тем, при сопоставлении данных термоанализа с величинами энергий стабилизации отдельных Н-связей (Естаб, кДж/моль) и величинами зарядов (qстаб, ед. з.), перенесенных в ходе их образования, такое согласие отсутствует. Это связано с тем, что величина Eint отражает суммарную энергетику взаимодействия “порфириноид–кислота”, тогда как величина Естаб – энергию отдельных орбитальных взаимодействий. Реальное же КОВ порфириноидов, согласно расчетным данным, обычно является многоцентровым, особенно, в случае корролов (табл. 2, рис. 4).
Рис. 4.
Оптимизированные структуры катионных солей II с одной молекулой НОАс (а) и двумя молекулами TFA (б), а также III с молекулой TFA (в).
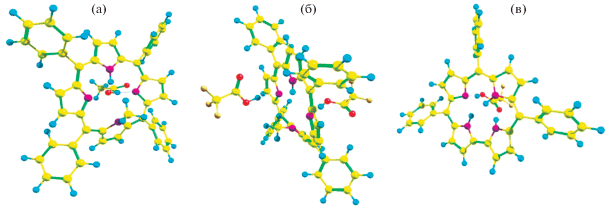
Результаты квантово-химических исследований показывают, что некоторые стадии протонирования N-основных центров порфириноидов, фиксируемые в растворе [21, 39], могут не наблюдаться в расчетных данных, полученных для идеализированных условий, в тех случаях, когда требуемые затраты энергии на конформационную или π-электронную перестройку макроцикла не компенсируются за счет образования новых связей.
Так, например, слабый протонодонор АсОН не взаимодействует со вторым N-основным центром монокатиона N-замещенной молекулы II, закрытым с одной стороны плоскости макроцикла метильной группой и молекулой кислоты – с другой (рис. 4а). При этом более сильная кислота (TFA) образует с этим макроциклом двукратнопротонированную форму (рис. 4б).
Второй пример – отсутствие C-протонирования монокатиона коррола III по мезо-углеродному атому в условиях квантово-химического расчета. Эти результаты согласуются с экспериментальными данными, полученными термогравиметрическим методом (табл. 1).
Согласно расчетным данным, одним из вероятных продуктов взаимодействия коррола (III) с первой молекулой карбоновой кислоты являются сложные Н-связанные мостиковые структуры, которые, вероятно, в растворе не реализуются и в которых N-центр является акцептором, а NH-центр – донором протона в отношении кислоты НХ (рис. 4в). Возникающие при этом водородные связи характеризуются достаточно высокой величиной Естаб, равной 93 и 78 кДж/моль в случае АсОН и TFA, соответственно (табл. 2). Наличие взаимодействия Н–Х…(Н–N) даже в составе протонированной формы макроцикла свидетельствует о высокой химической активности NH-связей молекул исходного H3Cor (III).
В случае таутомера а инвертированного порфириноида (IV) существуют два альтернативных N-центра протонирования – внешний и внутренний. Как и следовало ожидать, образование монокатиона с участием внешнециклического N-атома является, согласно данным NBO-анализа, более благоприятным. Так, энергия взаимодействия протона кислоты (TFA) с внешним N-атомом или, иными словами, энергия стабилизации водородной связи (Естаб), 308 кДж/моль) значительно, на 231 кДж/моль, выше по сравнению с внутрициклическим, а величина переноса заряда (0.272 ед.з.) – на 0.23 ед.з. выше. Как правило, рост величин Естаб и qстаб сопровождается уменьшение расстояний (rX ↔ Y–H, rX ↔ H, табл. 2) между взаимодействующими центрами.
Образование протонированных форм макрогетероциклов I–IV приводит к существенному искажению плоской структуры, что следует из величин двугранных углов между парами Сβ-атомов (φβ–β), атомов азота МГЦ (φN–N), а также углов вращения мезо-фенильных колец (φAr, табл. 2). Дипольные моменты в ходе протонирования МГЦ также существенно возрастают за счет поляризации молекулы, особенно, в случае присоединения протона к внешнециклическому N-атому IVа или единственному третичному N-атому в молекуле коррола III (табл. 2). Образование симметричной дикатионной формы Н(n + 2)Р2+, как в случае I, приводит к полной деполяризации молекулы (μ ∼ 0, табл. 2).
Таким образом, анализ приведенных выше экспериментальных результатов позволяет сделать следующие выводы:
1. Катионные соли порфирина (I) и порфириноидов (II–IV) в инертной среде во всех случаях подвергаются термическому разложению до свободного лиганда НnP, одной или двух молекул связанной с ним кислоты и от 4 до 15 молекул воды; число молекул варьирует в зависимости от природы НХ и протонированного макрогетероцикла.
2. Моделирование структуры катионных форм соединений I–IV в идеальной газовой фазе посредством квантово-химических расчетов показало, что взаимодействие лиганда НnP с одной или двумя молекулами кислоты ограничивается образованием Н-связей с высокой степенью переноса заряда до 0.27 ед. Изменения энергии взаимодействия “макрогетероцикл–кислота” (ΔЕint), характеризующие основность МГЦ I–IV, согласуются с величинами энтальпий испарения молекул кислот из солей порфириния, рассчитанными из термогравиметрических данных.
Работа выполнена при частичной поддержке Российского научного фонда (соглашение № 18-73-00217). Исследование проведено с использованием ресурсов Центра коллективного пользования научным оборудованием ФГБОУ ВО “ИГХТУ”.
Список литературы
The porphyrin handbook / Ed. by K.M. Kadish, K.M. Smith, R. Guilard. New York, CA: Acad. Press, 2000. V. 2. 420 p.
Mack J., Kobayashi N., Shen Z. Handbook of Porphyrin Science / Ed. by K.M. Kadish, K.M. Smith, R. Guilard. Acad. Press, 2013. V. 23. P. 109, 281–371.
Paolesse R. Applications of Porphyrinoids. Springer, 2014. 184 p.
Kustov A.V., Berezin M.B. // J. Chem. Eng. Data. 2013. V. 58. № 9. P. 2502. https://doi.org/10.1021/je400388j
Kustov A.V., Smirnova N.L., Berezin D.B., Berezin M.B. // J. Chem. Thermodyn. 2015. V. 89. P. 123. https://doi.org/10.1016/j.jct.2015.05.016
Kustov A.V., Smirnova N.L., Berezin D.B., Berezin M.B. // Ibid. 2015. V. 83. P. 104–109. https://doi.org/10.1016/j.jct.2014.12.013
Андрианов В.Г., Березин Д.Б., Малкова О.В. // В кн.: Успехи химии порфиринов. СПб.: Изд. СПбГУ, 2001. Т. 3. С. 107; Andrianov V.G., Berezin D.B., Malkova O.V. // In: Uspekhi khimii porfirinov. St.-Petersburg: SPbGU ed., 2001. V. 3. P. 107–129 (in Russ.)
Rosa A., Ricciardi G., Baerends E.J. // J. Porph. Phthaloc. 2006. V. 10. № 4–6. P. 381.
Presselt M., Dehaen W., Maes W., Klamt A., Martinez T., Beenken W.J.D., Kruk M. // Phys. Chem. Chem. Phys. 2015. V. 17. P. 14096. https://doi.org/10.1039/C5CP01808K
Kustov A.V., Kruchin S.O., Smirnova N.L., Berezin D.B. // Macroheterocycles. 2016. V. 9. № 4. P. 373–377. https://doi.org/10.6060/mhc160647k
Березин Б.Д., Андрианов В.Г., Березин Д.Б. // Координац. химия. 1994. Т. 20. № 5. С. 350; Berezin B.D., Andrianov V.G., Berezin D.B. // Rus. J. Coord. Chem. 1994. V. 20. № 5. P. 350.
Березин Д.Б. // Дисс. докт. хим. наук. Иваново: ИГХТУ, 2007. 350 с.; Berezin D.B. // Doctoral thesis. Ivanovo: ISUCT, 2007. 350 p. (in Russ.)
Петров О.А. // Журн. общей химии. 2013. Т. 83. № 4. С. 681; Petrov O.A. // Russ. J. Gen. Chem. V. 83. № 6. С. 1136–1142. https://doi.org/10.1134/S1070363213060224
Berezin D.B., Karimov D.R. // Macroheterocycles. 2009. V. 2. № 1. P. 42–51.
Chirvony V.S., Van Hoek A., Galievsky V.A. et al. // J. Phys. Chem. 2000. V. 104. № 42. P. 9909. https://doi.org/10.1021/jp001631i
Березин Д.Б., Андрианов В.Г., Семейкин А.С., Березин М.Б. // Журн. общ. химии. 2000. Т. 70. В. 9. С. 1547; Berezin D.B., Andrianov V.G., Semeykin A.S., Berezin M.B. // Rus. J. Gen. Chem. 2000. V. 70. Iss. 9. P. 1547–1552.
Березин Д.Б., Каримов Д.Р., Березин М.Б. // Журн. физ. химии. 2013. Т. 87. № 4. С. 615–620; Berezin D.B., Karimov D.R., Berezin M.B. // Russ. J. Phys. Chem. A. 2013. V. 87. № 4. P. 593–597. https://doi.org/10.1134/S0036024413040055
Березин Д.Б., Крестьянинов М.А. // Журн. структур. химии. 2014. Т. 55. № 5. С. 868; Berezin D.B., Krest’yaninov M.A. // Rus. J. Struct. Chem. 2014. V. 55. № 5. P. 822–830. https://doi.org/10.1134/S0022476614050047
Березин Д.Б., Таланова А.Е., Крестьянинов М.А. и др. // Журн. физ. химии. 2016. Т. 90. № 10. С. 1465; Berezin D.B., Talanova A.E., Serov I.N., Semeikin A.S., Krest’yaninov M.A. // Russ. J. Phys. Chem. A. 2016. V. 90. № 10. P. 1948–1955. https://doi.org/10.1134/S0036024416100058
Березин Д.Б., Каримов Д.Р., Баранников В.П., Семейкин А.С. // Журн. физ. химии. 2011. Т. 85. № 12. С. 2325; Berezin D.B., Karimov D.R., Semeikin A.S., Barannikov V.P. // Russ. J. Phys. Chem. A. 2011. Т. 85. № 12. P. 2171–2176. https://doi.org/10.1134/S0036024411120041
Березин Д.Б., Каримов Д.Р., Кустов А.В. Корролы и их производные: синтез, свойства, перспективы практического применения. М.: Ленанд, 2018. 304 с. Berezin D.B., Karimov D.R., Kustov A.V. Corroles and their derivatives: synthesis, properties, possibility for practical application. M.: Lenand, 2018. 304 p. (in Russ.)
Березин Д.Б., Андрианов В.Г., Семейкин А.С. // Оптика и спектроскопия. 1996. Т. 80. № 4. С. 618; Berezin D.B., Andrianov V.G., Semeikin A.S. // Optics and Spectroscopy. 1996. V. 80. № 4. P. 618.
Latos-Grażyński L. // Inorg. Chem. 1985. V. 24. № 11. P. 1681. https://doi.org/10.1021/ic00205a018
Koszarna B., Gryko D.T. // J. Org. Chem. 2006. V. 71. № 10. P. 3707. https://doi.org/10.1021/jo060007k
Geier G.R., III, Lindsey J.S. // Org. Letters. 1999. V. 1. № 9. P. 1455. https://doi.org/10.1021/ol9910114
Бургер К. Сольватация, ионные реакции и комплексообразование в неводных средах / Пер. с англ. М.: Мир, 1984. 256 с.; Burger K. Solvation, ionic and complex formation reactions in non-aqueous solvents. Elsevier, 1983.
Barannikov V.P., Vyugin A.I., Antina E.V., Krestov G.A. // Thermochimica Acta. 1990. V. 169. P. 103.
Gaussian 09, Revision D.01, Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., J.J. Dannenberg, Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., and Fox D.J., Gaussian, Inc., Wallingford CT, 2009.
Ditchfield R., Hehre W.J., Pople J.A. // J. Chem. Phys. 1971. V. 54. P. 724.
Weinhold F. // J. Mol. Struct. (Theochem). 1997. V. 398–399. P. 181.
Glendening E.D., Reed A.E., Weinhold F. NBO 3.0 Program manual. University of Wisconsin. Madison, 1998.
Alabugin I.V., Manoharan M., Peabody S. Weinhold F. // J. Amer. Chem. Soc. 2003. V. 125. № 19. P. 5973. https://doi.org/10.1021/ja034656e
Sazanovich I.V., van Hoek A., Panarin A.Yu. et al. // J. Porph. Phthaloc. 2005. V. 9. № 1. P. 59-67; https://doi.org/10.1142/S1088424605000113
Березин Д.Б., Мальцев И.А., Семейкин А.С., Болотин В.Л. // Журн. физ. химии. 2005. Т. 79. № 12. С. 2220; Berezin D.B., Mal’tsev I.A., Semeykin A.S., Bolotin V.L. // Rus. J. Phys. Chem. 2005. V. 79. № 12. P. 2220–2226.
Березин Д.Б. N-замещенные порфириноиды: строение, спектроскопия, реакционная способность. LAP. Saarbrücken. 2012. 56 с.; Berezin D.B. N-substituted porphyrinoids: structure, spectroscopy, reactivity. LAP. Saarbrücken. 2012. 56 p. (in Russ.)
Sakashita R., Ishida M., Furuta H. // J. Phys. Chem. A. 2015. V. 119. № 6. P. 1013–1022. https://doi.org/10.1021/jp512229k
Sheinin V.B., Kulikova O.M., Aleksandriiskii V.V., Koifman O.I. // Macroheterocycles. 2016. V. 9. № 4. P. 353–360. https://doi.org/10.6060/mhc161067s
Березин Д.Б., Ву Тхи Тхао, Азорина А.А. и др. // Журн. неорган. химии. 2015. Т. 60. № 10. С. 1385; Berezin D.B., Vu Thi Thao, Azorina A.A., Shukhto O.V., Guseinov S.S., Berezina N.M. // Russ. J. Inorg. Chem. 2015. V. 60. № 10. P. 1267–1274. https://doi.org/10.1134/S0036023615100046
Березин Д.Б., Шухто О.В., Ву Тхи Тхао и др. // Журн. неорган. химии. 2014. Т. 59. №. 12. С. 1769; Berezin D.B., Shukhto O.V., Vu Thi Thao, Berezin B.D. // Russ. J. Inorg. Chem. 2014. V. 59. № 12. P. 1522−1529. https://doi.org/10.1134/S0036023614120067
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии