

Журнал физической химии, 2020, T. 94, № 10, стр. 1533-1538
Применение гидросиликагеля, выделенного из серпентинов, для получения наноразмерных кристаллов β-волластонита
А. Р. Исаакян a, *, Н. О. Зулумян a, С. А. Меликян a, А. А. Бегларян a, b
a Национальная академия наук Республики Армения, Институт общей и неорганической химии
0051 Ереван, Армения
b Ереванский государственный университет
0025 Ереван, Армения
* E-mail: Isahakyananna@yahoo.com
Поступила в редакцию 06.11.2019
После доработки 06.11.2019
Принята к публикации 14.04.2020
Аннотация
Исследовано взаимодействие гидроксида кальция Ca(OH)2 с гидросиликагелем, выделенным из серпентинов (Mg(Fe))6[Si4O10](OH)8. Установлено, что при использовании данного гидросиликагеля в качестве SiO2 удается избежать многочасовую автоклавную обработку и обычным перемешиванием в условиях атмосферного давления нагретой до температуры кипения суспензии, приготовленной из указанных реагентов, получить такие аморфные интермедианты, которые при отжиге в температурном интервале 800–850°C превращаются в наноразмерные кристаллы β-волластонита.
Новый подход к химической обработке серпентинитов11 позволил получить из серпентиновых минералов наряду с соединениями магния и железа(III) гидросиликагель, содержащий до 7% аморфного диоксида кремния SiO2 [1, 2]. По сути, гидросиликагель является конденсатом кремниевых кислот, образованных из различных силикатных анионов (орто- [SiO4]4–, ди-[Si2O7]6–, три- [Si3O10]8– и т.п.), в свою очередь, выделенных выщелачиванием из структуры серпентина [3, 4].
Недавние исследования показали, что данный гидросиликагель может не только успешно применяться в качестве источника кремнезема для получения силикатов стронция и бария при низких температурах, но и значительно упростить процедуру их получения методом осаждения в жидкой среде и разработать новыe методики, обеспечивающие синтез монофазы наноразмерных кристаллов ортосиликата стронция Sr2SiO4 (20–40 нм) и метасиликата бария (25–80 нм) [5–7].
Полученные результаты натолкнули на идею расширить область применения гидросиликагеля и изучить его взаимодействие с гидроксидом еще одного элемента второй подгруппы – кальцием Ca, что возможно также позволит получать из него силикаты кальция, в частности, β-волластонит (β-CaSiO3), более простым способом. Один из наиболее распространенных методов получения синтетического β-CaSiO3, основанный на отжиге промежуточной твердой фазы, предварительно полученной в жидкой среде путем взаимодействия гидроксида кальция Ca(OH)2 с кремнезем содержащими реагентами, весьма энергоемкий, поскольку требует автоклавные условия и многочасовую обработку [8–14].
Напомним, что β-волластонит – неорганический полимер, обладающий рядом параметров, таких как высокая химическая стойкость, низкая теплопроводность, игольчатый габитус частиц, высокая температура плавления, исключительная белизна, которые делают его экологически чистым, безопасным материалом многоцелевого назначения: в качестве сорбента, наполнителя, армирующей добавки в композиционных материалах, при производстве бумаги и в качестве пигмента [15–22].
В настоящей работе с помощью методов рентгенофазового (РФА), дифференциально-термического (ДТА) анализов и сканирующей электронной микроскопии (СЭМ) исследовалось взаимодействие Ca(OH)2 с гидросиликагелем, выделенным из серпентиновых минералов, и термическое превращение образуюшихся интермедиантов в силикаты кальция, в частности в β-волластонит.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве диоксида кремния использовали гидросиликагель (SiO2 5.8%), полученный по разработанной методике [2] из образца серпентинита, взятого с месторождения Шоржа (Армения).
Oксид кальция CaO 8677-76 Марки “ч.”, выдержанный 0.5 ч при температуре 1000°C, растворяли в дистиллированной воде для получения Ca(OH)2.
Четыре образца суспензий подготавливали из гидросиликагеля и раствора Ca(OH)2. В реакционный сосуд вносили навеску гидросиликагеля и добавляли Ca(OH)2, которые брали в мольных соотношениях SiO2 : CaO (для краткости далее S : C), равных 1 : 1, 1 : 2, 1 : 1.4, 1 : 1.6, и суспендировали их в дистиллированной воде при соотношении твердой и жидкой фаз (Т : Ж) 1 : 15. Затем каждый образец нагревали до температуры кипения (95°C) и перемешивали приводной мешалкой в течение 2 ч. Пульпу, образующуюся после обработки каждого образца, отделяли от раствора фильтрованием через бумажный фильтр и промывали дистиллированной водой. Полученный осадок выдерживали при температуре 60–80°C в течение 24 ч в сушильном шкафу KBC G-100/250 производства фирмы Premed (Варшава, Польша).
Каждый из высушенных образцов подвергали РФА и ДТА от комнатной температуры до 1000°C, а также РФА после ДТА. Масса исследуемых образцов составляла 300 мг для ДТА и 220–240 мг для РФА.
Один из высушенных образцов подвергали получасовой термообработке при различных температурах в диапазоне от 200 до 900°C. Еще один образец, демонстрирующий наилучшие результаты, был также выдержан 0.5 ч при 800 и 900°C. Значения температур обжига устанавливали и контролировали с помощью муфельной печи Wise Therm F digital (China). Затем каждый термообработанный образец исследовали с помощью РФА.
ДТА был проведен на дериватографе DERIVATOGRAPH Q-1500 D фирмы МОМ (Венгрия) в среде атмосферы со скоростью нагрева 10°C мин–1.
РФА осуществляли в CuΚα-излучении на дифрактометре ДРОН-3 (Россия) с использованием никелевого фильтра. Съемку проводили в интервале углов 2θ = 8°–80° при температуре 22°C на воздухе. Скорость движения счетчика 2 град мин–1. Все рефлексы отражения расшифрованы и идентифицированы с помощью базы данных JCPDS–ICDD 2004 года.
Средний размер частиц в конечном продукте был автоматически рассчитан с помощью программы Match!, используемой известную формулу Шеррера: L = 0.94λ/βcosθ, где β – полуширина линии; λ – длина волны; θ – угол дифракции; L – размер кристалла [23].
Микроструктурa синтезированного соединения была определена с помощью электронного микроскопа Tesla BS-300 (Чехия).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Поскольку рентгенограммы трех образцов, приготовленных из реагентов с мольным соотношением S : C 1 : 1, 1 : 2, 1 : 1.4 и высушенных при температуре 100°C, абсолютно одинаковы: на них прослеживаются только рефлексы карбоната кальция CaСO3 (Card № 83–0578), то в работе приводится дифрактограмма только одного образца с мольным соотношением S : C 1 : 1.4 (рис. 1). Дальнейшее увеличение концентрации Ca(OH)2 в первичном растворе до получения S : C, равного 1 : 1.6, приводит к появлению дополнительных рефлексов непрореагировавшего Ca(OH)2 (Card № 84–2041) (рис. 1).
Рис. 1.
Дифрактограммы образцов-интермедиантов, полученных из Ca(OH)2 и гидросиликагеля. Мольное соотношение S : C: 1 : 1.4, 1 : 1.6.

Независимо от мольного соотношения исходных веществ S : C на кривых ДТА всех четырех образцов в температурном интервале 100–800°C наблюдается ряд отличающихся по интенсивности эндотермических эффектов, и один интенсивный экзотермический эффект в области выше 800°C (рис. 2). Ход термогравиметрических (ТГ) и дифференциальных термогравиметрических (ДТГ) кривых показывает, что эндотермические явления сопровождаются потерей массы, а экзотермический процесс протекает без каких-либо массовых изменений (рис. 2).
Рис. 2.
Кривые ДТА образцов-интермедиантов, полученных из Ca(OH)2 и гидросиликагеля. Мольное соотношение S : C = 1 : 1, 1 : 1.2, 1 : 1.4, 1 : 1.6.

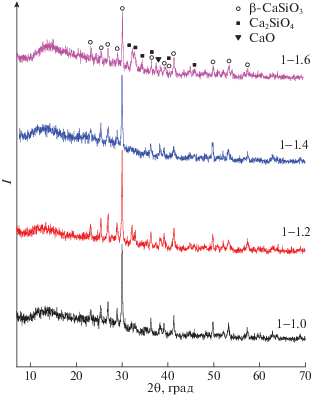
Согласно результатам рентгенофазового анализа образцов, полученных после ДТА, при нагреве до 1000°C промежуточные образцы кристаллизуются в силикаты кальция, а именно в β‑волластонит, а в некоторых случаях и в ларнит (Ca2SiO4) (рис. 3). Доказательством этого являются интенсивные диффракционные пики β-CaSiO3, прослеживаемые на дифрактограммах всех образцов (рис. 3). Из-за идентичности рефлексов отражения, свойственным полиморфным модификациям β-волластонита, моноклинного и триклинного (Card № 84–0654 и № 84–0655 соответственно), синтезируемый β-CaSiO3 можно считать или одной из форм этих модификаций, или их смесью [24]. Рефлексы моноклинного ларнита (Ca2SiO4) (Card № 83–0460) наряду с дифракционными пиками волластонита и оксида кальция CaO (Card № 77–2376) появляются лишь на дифрактограмме образца с мольным соотношением S : C 1 : 1.6 (рис. 3).
Рис. 3.
Дифрактограммы образцов, полученных после ДТА. Мольное соотношение S : C = 1 : 1, 1 : 1.2, 1 : 1.4, 1 : 1.6.
Для выяснения всех термически индуцированных процессов и фазовых превращений, протекающих в промежуточных соединениях при нагреве, был проведен детальный РФА одного из интермедиантов, полученного после термообработки при различных температурах в диапазоне от 200 до 900°C. В качестве объектов исследований был выбран промежуточный образец, синтезированный из исходных реагентов с мольным соотношением S : C 1 : 1.6.
При выдержке образца в температурном интервале 200–500°C на рентгенограмме кроме рефлексов отражения оксида кальция и карбоната кальция, интенсивность которых медленно падает при повышении температуры отжига, никакие дифракционные пики не регистрируются (рис. 4). Термообработка до 700°C приводит к полному разложению карбоната кальция и началу формирования силикатов кальция, на что указывает исчезновение рефлексов отражения, свойственных карбонату кальция, и появление слабых, едва заметных дифракционных пиков, характерных для β-CaSiO3 и Ca2SiO4 (рис. 4). Дальнейший нагрев образца до 900°C уже приводит к возрастанию их интенсивностей, особенно для β-волластонита (рис. 4).
Рис. 4.
Дифрактограммы термообработанного при различных температурах интермедианта, полученного из Ca(OH)2 и гидросиликагеля с мольным соотношением S : C = 1 : 1.6.

Эндотермические эффекты, наблюдаемые на кривых ДТА в области низких температур (100–210°C), вызваны удалением из синтезированных промежуточных веществ, адсорбированной и кристаллической воды, что косвенно указывает на образование гидросиликата кальция (рис. 2). Эндотермический пик с минимумом при 493°C, прослеживаемый на кривой ДТА образца с S : C 1 : 1.6 (рис. 2), обусловлен процессом разложения непрореагировавшего избытка гидроксида кальция на воду и оксид кальция, рефлексы которого прослеживаются на соответствующих рентгенограммах (рис. 3). Из РФА следует, что эндотермический эффект, едва заметный на кривых ДТА в области 600–700°C, вызван процессом разложения карбоната кальция. Следующий эндотермический процесс в температурном интервале в 700–800°C, скорее всего обусловленный процессом дегидроксилации гидроксосиликатов кальция, образующихся при перемешивании, плавно переходит в яркий экзотермический пик в области 810–850°C (рис. 2). Наиболее стремительный интенсивный экзотермический эффект наблюдается для образца с S : C 1 : 1.4. Сопоставление результатов РФА с ДТА указывает на то, что экзотермические пики данных образцов вызваны процессом образования силикатов кальция. Для образцов с мольным соотношением S : C 1 : 1, 1 : 1.2 и 1 : 1.4 они, главным образом, обусловлены формированием кристаллов β-волластонита, а с S : C 1 : 1.6 – также и ларнита, который образуется немного раньше, при более низких температурах, о чем свидетельствует экзотермический эффект с максимумом при 825°C, едва прослеживаемый в виде плеча (рис. 2). Отметим, что гидроксосиликаты кальция не обнаруживаются не рентгенограмме, поскольку находятся в аморфном состоянии.
Рефлексы одинаковой интенсивности регистрируются на дифрактограмме интермедианта с S : C 1 : 1.4, выдержанного 0.5 ч при 800 и 900°C (рис. 5) . Согласно уравнению Шеррера средний размер синтезируемых частиц составляет 80–100 нм.
Рис. 5.
Дифрактограммы термообработанного в течение 0.5 ч при 800 и 900°С образца, полученного из Ca(OH)2 и гидросиликагеля с мольным соотношением S : C = 1 : 1.4.

Исследование морфологии образца с S : C 1 : 1.4, полученного после термообработки при 800°C, показало, что синтезируемые кристаллы β-волластонита отличаются монодисперсностью (рис. 6).
Рис. 6.
Микрофотография конечного образца, синтезированного термообработкой в течение 0.5 ч при 800°C интермедианта, приготовленного из Ca(OH)2 и гидросиликагеля с мольным соотношением S : C = = 1 : 1.4.

Из экспериментальных данных следует, что в процессе перемешивания исходных реагентов, взятых в мольных соотношениям S : C 1 : 1–1.4, Ca(OH)2 реагирует с олигомерными силикатными анионами, составляющими данный гидросиликагель, образуя аморфные гидроксосиликаты кальция, которые при нагреве до 800°C кристаллизуются в β-CaSiO3. Увеличение концентрации Ca(OH)2 до получения отношения S : C, равного 1 : 1.6, способствует дальнейшему разделению этих олигомерных анионов до ортосиликатных единиц и формированию таких промежуточных гироксосиликатов кальция, из которых наряду с β-волластонитом формируется ларнит. Оптимальное соотношение, обеспечивающее высокий выход β‑CaSiO3 – S : C = 1 : 1.4.
Итак, гидросиликагель, выделенный из серпентинового минерала, может не только успешно применяться в качестве кремнезем содержащего реагента для получения наноразмерного β-волластонита, но и позволяет значительно упростить технологию его получения, исключая автоклавную обработку и сокращая длительность синтеза и термообработки. В результате обычного двухчасового перемешивания в условиях атмосферного давления кипящей водной суспензии, приготовленной из данного гидросиликагеля и Ca(OH)2, взятых в мольном соотношении S : C 1 : 1.4, образуется такой гидроксосиликат кальция, получасовая термообработка которого при 800°C оказывается достаточной для формирования наноразмерных кристаллов β-волластонита.
Список литературы
Zulumyan N.O., Isaakyan A.R., Oganesyan Z.G. // Russ. J. Appl. Chem. 2007. V. 80. № 6. P. 1020. https://doi.org/10.1134/S1070427207060353
Пат. РФ 2407704 C2 (опубл. 2010). Способ комплексной обработки серпентинитов.
Zulumyan N., Mirgorodski A., Isahakyan A., Beglaryan H. // J. Therm. Anal. Calorim. 2014. V. 115. № 2. P. 1003. https://doi.org/10.1007/s10973-013-3483-7
Zulumyan N., Isahakyan A., Beglaryan H., Melikyan S. // J. Therm. Anal. Calorim. 2018. V. 131. № 2. P. 1201. https://doi.org/10.1007/s10973-017-6705-6
Beglaryan H.A., Melikyan S.A., Terzyan A.M. et al. // Rus. J. Inorg. Chem. 2018. V. 63. № 11. P. 1395. https://doi.org/10.1134/s0036023618110025
Zulumyan N., Isahakyan A., Beglaryan H. et al. // J. Therm. Anal. Calorim. 2019. https://doi.org/10.1007/s10973-019-08035-9
Beglaryan H.A., Melikyan S.A., Zulumyan N.H. et al. // J. Mol. Liq. 2019. V. 291. P. 111263. https://doi.org/10.1016/j.molliq.2019.111263
Ismail H., Shamsudin R., Abdul Hamid M.A. // Materials Science & Engineering. C, Materials for biological applications. 2016. V. 58. P. 1077. https://doi.org/10.1016/j.msec.2015.09.030
Wu H., Yang J., Ma H.W., Wang M.W. // Integrated Ferroelectrics. 2013. V. 146. № 1. P. 144. https://doi.org/10.1080/10584587.2013.789777
Yazdani A., Rezaie H.R., Ghassai H., Mahmoudian M. // J. Ceram. Process. Res. 2013. V. 14. № 1. P. 12.
Lin K., Chang J., Liu X., Ning C. // International Journal of Applied Ceramic Technology. 2010. V. 7. № 2. P. 178. https://doi.org/10.1111/j.1744-7402.2009.02474.x
Grigoryan K.G., Arutunyan G.A., Baginova L.G., Grigoryan G.O. // Theor. Found. Chem. Eng. 2008. V. 42. № 5. P. 583. https://doi.org/10.1134/s0040579508050163
Lin K., Chang J., Chen G. et al. // J. Cryst. Growth. 2007. V. 300. № 2. P. 267. https://doi.org/10.1016/j.jcrysgro.2006.11.215
Lin K., Chang J., Lu J. // Mater. Lett. 2006. V. 60. № 24. P. 3007. https://doi.org/10.1016/j.matlet.2006.02.034
Садрашева А. // Ползуновский Альманах 2016. № 3. С. 189.
Гордиенко П.С., Ярусова С.Б., Козин А.В. и др. // Перспективные материалы. 2017. № 9. С. 40.
Ding Q., Zhang Z., Wang C. et al. // J. Therm. Anal. Calorim. 2014. V. 115. № 1. P. 675. https://doi.org/10.1007/s10973-013-3171-7
Morsy R., Abuelkhair R., Elnimr T. // Silicon. 2014. P. 1. https://doi.org/10.1007/s12633-014-9243-x
Sreekanth Chakradhar R.P., Nagabhushana B.M., Chandrappa G.T. et al. // Mater. Chem. Phys. 2006. V. 95. № 1. P. 169. https://doi.org/10.1016/j.matchemphys.2005.06.002
Yun Y.-H., Yoon C.-H., Kim Y.-H. et al. // Ceram. Int. 2002. V. 28. № 5. P. 503. https://doi.org/10.1016/S0272-8842(02)00002-0
Negmatov N.S., Abdullaev Z.Z. // Glass Ceram. 2001. V. 58. № 11. P. 396. https://doi.org/10.1023/a:1014914526841
Kokubo T. // Biomaterials. 1991. V. 12. № 2. P. 155. https://doi.org/10.1016/0142-9612(91)90194-F
Langford J.I., Wilson A.J.C. // J. Appl. Crystallogr. 1978. V. 11. № 2. P. 102. https://doi.org/10.1107/S0021889878012844
Tolliday J. // Nature. 1958. V. 182. № 4641. P. 1012. https://doi.org/10.1038/1821012a0
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии