

Журнал физической химии, 2020, T. 94, № 10, стр. 1490-1495
Влияние смешанного растворителя вода–ацетон на устойчивость глицилглицинатных комплексов меди(II)
В. А. Исаева a, *, А. С. Молчанов b, К. А. Кипятков a, К. В. Граждан a, Е. С. Розанов a
a Ивановский государственный химико-технологический университет
Иваново, Россия
b Костромской государственный университет
Кострома, Россия
* E-mail: kvol1969@gmail.com
Поступила в редакцию 28.11.2019
После доработки 28.11.2019
Принята к публикации 21.01.2020
Аннотация
Методом потенциометрического титрования при температуре 298 K и ионной силе растворов 0.1 (NaClO4) определены константы устойчивости нормальных и депротонированных моно- и бис-глицилглицинатных комплексов меди(II) в растворителе вода–ацетон переменного состава. На основе полученных данных рассчитаны константы диссоциации пептидной группы глицилглицинатного комплекса меди(II) в водно-ацетоновых смесях. Установлено, что с увеличением концентрации ацетона в растворе устойчивость всех комплексов меди(II) с глицилглицинат-ионом возрастает. Дана оценка вкладов пересольватации реагентов в водно-ацетоновом растворителе в изменение энергии Гиббса реакции образования глицилглицината меди(II). Установлено, что рост константы устойчивости глицилглицинатного комплекса меди(II) в водно-ацетоновом растворе определяется ослаблением сольватации лиганда. Проведено сравнение полученных результатов с аналогичными данными для реакций комплексообразования глицилглицинат-иона с ионами меди(II) и никеля(II) в водных растворах этанола и диметилсульфоксида.
Проведенные ранее исследования влияния состава и природы смешанного растворителя на устойчивость комплексов d-металлов с простейшими заряженными лигандами карбоксилатного типа (ацетат-ионом, глицинат-ионом) выявили закономерности в изменении термодинамических параметров реакций комплексообразования и сольватации реагентов [1]. Изучение процессов комплексообразования глицилглицинат-иона с ионом никеля(II) в различных водно-органических растворителях [2–4], а также с ионом меди(II) в растворителях вода–этанол [5] и вода–диметилсульфоксид [6] показало некоторые особенности в изменении термодинамических параметров этих реакций. В продолжение этих исследований в данной работе изучено влияние состава водно-ацетонового растворителя на устойчивость глицилглицинтных комплексов меди(II).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Константы устойчивости глицилглицинатных комплексов меди(II) определяли при температуре 298 K потенциометрическим методом с использованием хлорсеребряного электрода сравнения, внутренний раствор которого был однороден по составу с исследуемым для уменьшения диффузионного потенциала, и индикаторного стеклянного электрода, на работоспособность которого в водно-ацетоновых смесях указывается в работе [7]. Титрантом служил раствор глицилглицината натрия (5 × 10–1 моль/л). Дозировку титранта осуществляли весовым способом с помощью микрошприца. В потенциометрическую ячейку помещали раствор, содержащий Cu(ClO4)2 (8 × 10–3 моль/л) и HClO4 (1 × 10–3 моль/л). Ионную силу раствора (µ = 0.1) поддерживали с помощью добавок перхлората натрия. Стандартный раствор по ионной силе и концентрации органического сорастворителя был однороден с исследуемым.
Перхлорат меди(II) получали из основного карбоната меди(II) (“х.ч.”) и хлорной кислоты (“х.ч.”) с последующей перекристаллизацией. Концентрацию раствора Cu(ClO4)2 определяли титрованием этилендиаминтетраацетатом натрия (ЭДТА). Перхлорат натрия марки “ч.” подвергали перекристаллизации из водного раствора с последующим высушиванием до постоянной массы. Раствор глицилглицината натрия готовили по точным навескам эквимолярных количеств глицилглицина (фирмы “Sigma” с содержанием основного вещества ≥99%) и бескарбонатного насыщенного раствора NaOH (“х.ч.”). Ацетон (“х.ч.”) использовали без дополнительной очистки.
Расчет констант устойчивости комплексов по результатам потенциометрического титрования проводили по программе PHMETR [8]. Погрешность численных значений констант изучаемых равновесных процессов оценивали на основе статистической обработки результатов 3–5 параллельных титрований. Условия проведения потенциометрического эксперимента были оптимальными для надежного определения константы образования нормального моноглицилглицинатного комплекса меди(II). Образование прочих комплексных частиц также характеризовалось достаточным выходом, однако ввиду высокой корреляции их констант устойчивости, определяемые значения lg β4, lg K5, lg K6 имеют более высокую погрешность.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Глицилглицинат-ион (GG–) проявляет комплексообразующие свойства с большинством d-металлов за счет наличия трех потенциально возможных центров координации: азота концевой аминогруппы, кислорода карбоксилатной группы и атома кислорода или азота пептидной группы. Процесс образования глицилглицинатов меди(II) сопровождается диссоциацией пептидной группы лиганда, приводя к образованию не только нормальных ([CuGG]+, [CuGG2]), но и депротонированных ([CuGG–H], [CuGG–HGG]–) глицилглицинатных комплексов Cu(II) [9, 10]. Установлено, что с ионом двухвалентной меди анион глицилглицина образует комплексы посредством азота аминогруппы и кислорода пептидной группы, координация по пептидному азоту в комплексах, образованных с недиссоциированным по пептидной группе глицилглицинат-ионом, не подтверждена [9, 10]. В образовании комплексов Cu(II) с депротонированным по пептидной группе глицилглицинат-ионом участвуют атомы азота амино- и пептидной групп лиганда [9, 10].
При расчете констант равновесий по данным потенциометрического титрования в водно-ацетоновом растворителе по программе PHMETR [8] учитывали протекание следующих реакций:
Адекватность математической модели реальным процессам проверялась контрольными расчетами по программе PHMETR [8] с введением в расчетную модель процессов образования [CuНGG]2+, [CuGG–HOH]– либо [CuOH]+, константы равновесия которых для водного раствора взяты из [13], [14] и [15] соответственно. Проверочные расчеты характеризовались ухудшением описания системы ввиду нечувствительности критериальной функции PHMETR [8] к этим формам. Подтверждение адекватности выбранной модели и отсутствия в ней неучтенных процессов – хорошая сходимость результатов обработки кривых титрования, снятых в водном растворе при различных соотношениях начальных концентраций лиганда, иона металла и протона, а также соответствие полученных констант для водного раствора литературным данным (табл. 1), полученным при аналогичных условиях (Т = 298 K, µ = 0.1 M). Хорошо соответствует литературным данным для водного раствора и значение константы диссоциации пептидной группы глицилглицината меди(II), рассчитанное по уравнению:
(1)
$\lg {{K}_{{\text{а}}}} = --{\text{р}}{{K}_{{\text{а}}}} = \lg {{\beta }_{4}}--\lg {{K}_{3}}{\kern 1pt} .$Таблица 1.
Константы образования глицилглицинатных комплексов меди(II) и депротонирования пептидной группы глицилглицината меди(II) (рKа) в водном растворе, Т = 298 K, µ = 0.1 М
| lg K3 [CuGG]+ | lg β4 [CuGG–H] | lg K5 [CuGG–HGG]– | рKа | Фоновый электролит | Источник |
|---|---|---|---|---|---|
| 5.56 | 1.46 | 3.18 | 4.10 | NaClO4 | наши данные |
| 5.68 | 1.47 | 2.84 | 4.21 | КNO3 | [13] |
| 5.56 | 1.50 | – | 4.06 | NaClO4 | [14] |
| 5.40 | 1.47 | 3.3 | 3.93 | КCl | [16] |
| 5.44 | 1.25 | – | 4.19 | КCl | [17] |
| 5.55 | 1.56 | – | 3.99 | NaClO4 | [18] |
| 5.71 | 1.56 | – | 4.15 | NaClO4 | [19] |
| 5.68 | 1.50 | – | 4.18 | КNO3 | [20] |
| 5.56 | 1.44 | 3.17 | 4.12 | КNO3 | [21] |
| 5.43 | 1.26 | – | 4.17 | NaCl | [22] |
Константа образования в водном растворе нормального бис-глицилглицинатного комплекса меди(II) lg K6 = 5.44, полученная нами при Т = = 298 K, µ = 0.1(NaClO4), с учетом различий в условиях эксперимента удовлетворительно соотносится с lg K6 = 5.17 (Т = 298 K, µ = 0.0) [23] и со значением lg K6 = 5.02 (Т = 293 K, µ = 0.2(KCl)), рассчитанным нами, исходя из константы диссоциации глицилглицина [11] и из брутто-констант, приведенных в [24] для реакций:
Таблица 2.
Константы образования глицилглицинатных комплексов меди(II) и депротонирования пептидной группы глицилглицината меди(II) (рKа) в водно-ацетоновом растворителе, Т = 298 K, µ = 0.1 (NaClO4)
| Константа равновесия |
Комплексная частица |
Состав растворителя, мол. доли ацетона | ||||
|---|---|---|---|---|---|---|
| 0.0 | 0.1 | 0.2 | 0.3 | 0.4 | ||
| lg K3 | [CuGG]+ | 5.56 ± 0.05 | 6.07 ± 0.05 | 6.68 ± 0.05 | 7.38 ± 0.05 | 7.82 ± 0.05 |
| lg β4 | [CuGG-H] | 1.46 ± 0.08 | 1.83 ± 0.09 | 2.37 ± 0.12 | 2.65 ± 0.15 | 2.85 ± 0.15 |
| lg K5 | [CuGG-HGG]¯ | 3.18 ± 0.08 | 4.44 ± 0.09 | 5.10 ± 0.13 | 5.98 ± 0.17 | 6.33 ± 0.17 |
| lg K6 | [CuGG2] | 5.44 ± 0.08 | 5.85 ± 0.09 | 6.04 ± 0.11 | 6.54 ± 0.11 | 6.77 ± 0.11 |
| рKa | 4.10 ± 0.08 | 4.24 ± 0.09 | 4.31 ± 0.15 | 4.73 ± 0.18 | 4.97 ± 0.18 | |
Рис. 1.
Константы депротонирования пептидной группы глицилглицината меди(II) в водно-органических растворителях (298 K, μ = 0.1 M (NaClO4)): 1 – вода–диметилсульфоксид, 2 – вода–этанол, 3 – вода–ацетон.
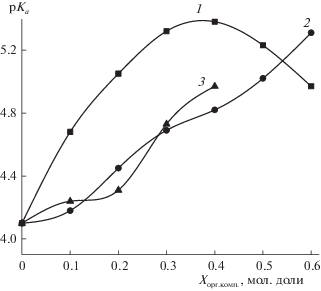
Согласно данным табл. 2, увеличение содержания в растворе ацетона способствует повышению устойчивости всех глицилглицинатных комплексов меди(II). Сопоставление полученных значений констант устойчивости глицилглицинатов меди(II) с аналогичными данными для других водно-органических растворителей показывает, что в водно-ацетоновых и водно-этанольных [5] растворах с увеличением концентрации неводного компонента наблюдается близкий к прямолинейному рост ступенчатых констант устойчивости, в водно-диметилсульфоксидных растворах [6] наибольший прирост констант устойчивости образующихся комплексов наблюдается в области составов растворителя 0.0–0.3 мол. доли, далее темпы роста lg Kуст замедляются (рис. 2 и 3). Аналогичный характер изменения констант устойчивости в данных водно-органических растворителях наблюдается для глицилглицинатных комплексов никеля(II) [2–4] (рис. 3).
Рис. 2.
Влияние водно-ацетонового (а), водно-диметилсульфоксидного (б), и водно-этанольного (в) растворителей на константы устойчивости глицилглицинатных комплексов меди(II) (298 K, μ = 0.1 M (NaClO4)): 1 – lg K6, 2 – lg K5, 3 – lg β4.

Рис. 3.
Влияние водно-ацетонового (а), водно-диметилсульфоксидного (б), и водно-этанольного (в) растворителей на константы устойчивости (lg K3) нормальных моноглицилглицинатных комплексов (298 K, μ = 0.1 M (NaClO4)): 1 – меди(II), 2 – никеля(II).
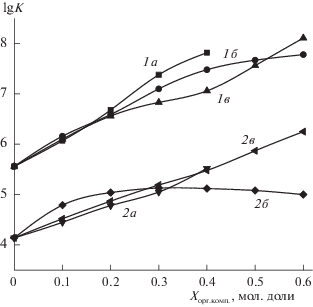
Сопоставление констант устойчивости нормального глицилглицинатного комплекса меди(II) в водно-ацетоновом растворителе с константами устойчивости комплексов меди(II) состава 1 : 1 с другими N-, O-донорными лигандами показывает, что для заряженных лигандов (ацетат-иона [25], глицинат-иона [26], глицилглицинат-иона) в водных растворах ацетона наблюдается рост устойчивости комплексов ∼2 лог. ед., меньший прирост lg Kуст (∼1 лог. ед.) наблюдается для комплексов меди(II) с пиридином [27] и его производным – никотинамидом [28], зависимость константы устойчивости аммиаката меди(II) от состава водно-ацетонового растворителя [1] носит слабовыраженный характер (рис. 4). Образование комплекса меди(II) с этилендиамином в водно-ацетоновом растворителе характеризуется уменьшением константы устойчивости [29] (рис. 4), поскольку при комплексообразовании d-металлов с этилендиамином в водных растворах ацетона константы устойчивости, рассчитанные из экспериментальных данных, представляют собой брутто-величину, включающую в себя вклады как взаимодействия металла с лигандом, так и взаимодействия амина с ацетоном [30].
Рис. 4.
Влияние водно-ацетонового растворителя на устойчивость монолигандных комплексов меди(II) (Т = = 298 K) с: 1 – этилендиамином (μ = 0.35 M (NaClO4)), 2 – глицинат-ионом (μ = 0.1 M (NaClO4)), 3 – глицилглицинат-ионом (μ = 0.1 M (NaClO4)), 4 – аммиаком (μ = 0.3 M (NaClO4)), 5 – ацетат-ионом (μ = 0.3 M (NaClO4)), 6 – пиридином (μ = 0.1 M (NaClO4)), 7 – никотинамидом (μ = 0.25 M (NaClO4)).

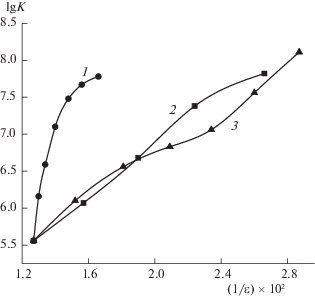
Рассмотрение причин изменения устойчивости комплекса меди(II) с глицилглицинат-ионом в водно-органических растворителях в рамках континуального подхода показывает, что изменение диэлектрических свойств смешанного растворителя оказывает значительное влияние на смещение равновесия реакции комплексообразования, однако определяющей причиной изменения устойчивости глицилглицината меди(II) в растворе не является. С ростом концентрации неводного компонента в растворе расхождение между значениями lg K3 для изодиэлектрических сред становится все более существенным, а в водно-диметилсульфоксидном растворителе линейная корреляция lg K3 от величины обратной диэлектрической проницаемости (1/ε) вообще отсутствует (рис. 5).
Рис. 5.
Зависимости константы устойчивости нормального моноглицилглицинатного комплекса меди(II) от диэлектрической проницаемости (ε) растворителей: 1 – вода–диметилсульфоксид, 2 – вода–ацетон, 3 – вода–этанол.
Рассмотрение сольватационных вкладов реагентов в смещение равновесия процесса образования нормального моноглицилглицинатного комплекса меди(II) в растворителе вода–ацетон, показывает (рис. 6), что изменение энергии Гиббса пересольватации иона Cu2+ в водно-ацетоновых смесях [31] способствует упрочнению комплекса. Отсутствие данных о пересольватации глицилглицинат-иона и комплекса [CuGG]+ в водно-ацетоновом растворителе позволяет рассчитать только величину различий в изменении ΔtrG° этих частиц (рис. 6), исходя из уравнения:
(2)
$\begin{gathered} {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r3}}}}^{^\circ } = ({{\Delta }_{{{\text{tr}}}}}G^\circ ({{[{\text{CuGG}}]}^{ + }})--{{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{G}}{{{\text{G}}}^{{\text{ - }}}})) - \\ --{{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{C}}{{{\text{u}}}^{{2 + }}}). \\ \end{gathered} $Рис. 6.
Изменение энергии Гиббса реакции образования нормального моноглицилглицината меди(II) и пересольватации реагентов в растворителе вода–ацетон: 1 – ΔtrG°(Сu2+), 2 – (ΔtrG°([CuGG]+) – ΔtrG°(GG–)), 3 – ΔtrGr3, 4 – ΔtrG° (GG–), рассчитанное при αdif = 0.5.
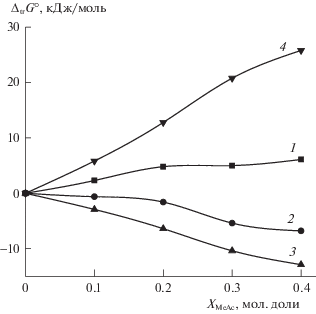
Оценить значение энергии Гиббса пересольватации глицилглицинат-иона в водно-ацетоновом растворителе можно, используя величину коэффициента различий αdif, который определяет долю ΔtrG° реакции комплексообразования от ΔtrG° лиганда [1]:
(3)
${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r3}}}}^{^\circ } = ({{\alpha }_{{{\text{dif}}}}}--1){{\Delta }_{{{\text{tr}}}}}G^\circ ({\text{G}}{{{\text{G}}}^{--}}).$Список литературы
Шарнин В.А. // Изв. вузов. Химия и хим. технология. 2005. Т. 48. Вып. 7. С. 44.
Наумов В.В., Исаева В.А., Ковалева Ю.А., Шарнин В.А. // Журн. физ. химии. 2013. Т. 87. № 7. С. 1160.
Исаева В.А., Наумов В.В., Шарнин В.А. // Коорд. химия. 2009. Т. 35. № 11. С. 878.
Наумов В.В., Исаева В.А., Шарнин В.А. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1208.
Исаева В.А., Молчанов А.С., Кипятков К.А., Шарнин В.А. // Журн. физ. химии. 2019. Т. 93. № 8. С.1164.
Исаева В.А., Молчанов А.С., Кипятков К.А., Шарнин В.А. // Там же. 2020. Т. 94. № 2. С. 182.
Батлер Дж. Электроды сравнения в апротонных органических растворителях, в кн.: Электрохимия металлов в неводных растворах / Под ред. Я.М. Колотыркина. М.: Мир, 1977. 440 с.
Бородин В.А., Козловский Е.В., Васильев В.П. // Журн. неорган. химии. 1986. Т. 31. № 1. С. 10.
Datta S.P., Rabin B.R. // Trans. Faraday Soc. 1956. V. 52. P. 1130.
Nakon R., Angelici R.J. // Inorg. Chem. 1973. V. 12. № 6. P. 1269.
Исаева В.А., Наумов В.В., Гессе Ж.Ф., Шарнин В.А. // Журн. физ. химии. 2009. Т. 83. № 3. С. 477.
Woolej E.H., Hurkot D.G., Herber L.G. // J. Phys. Chem. 1970. V. 74. № 22. P. 3908.
Kaneda A., Martell A. // J. Coord. Chem. 1974. V. 4. P. 137.
Brunetti A., Lim M., Nancollas G. // J. Am. Chem. Soc. 1968. V. 90. P. 5120.
Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1971. 456 с.
Bordignon-Luiz M., Szpoganicz B., Rizzoto M. et al. // Inorg. Chim. Acta. 1997. V. 254. P. 345.
Яцимирский К.Б., Манорик П.А., Давиденко Н.К. // Коорд. химия. 1988. Т. 14. № 3. С. 311.
Sigel H., Prijs B., Martin R. // Inorg. Chim. Acta. 1981. V. 56. P. 45.
Sigel H., Grisser R., Prijs B. // Z. Naturforsch. 1972. V. 27B. P. 353.
Yamauchi O., Hirano Y., Nakao Y., Nakahara A. // Can. J. Chem. 1969. V. 47. P. 3441.
Martin R. // Bull. Soc. Chim. Fr. 1967. P. 2217.
Biester J.L., Ruoff P. M. // J. Am. Chem. Soc. 1959. V. 81. P. 6517.
Кочергина Л.А., Емельянов А.В. // Журн. физ. химии. 2015. Т. 89. № 4. С. 592.
Kittl W., Rode B. // Inorg. Chim. Acta. 1981. V. 55. P. 21.
Исаева В.А., Шарнин В.А., Шорманов В.А., Щербина И.В. // Коорд. химия. 1998. Т. 24. № 2. С. 149.
Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Журн. неорган. химии. 1997. Т. 42. № 7. С. 1220.
Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Там же. 1997. Т. 42. № 7. С. 1224.
Исаева В.А., Гессе Ж.Ф., Шарнин В.А. // Коорд. химия. 2006. Т. 32. № 5. С. 340.
Михеев С.В., Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Журн. неорган. химии. 1994. Т. 39. № 9. С. 1502.
Шарнин В.А. // Журн. общ. химии. 1994. Т. 64. Вып. 11. С. 1914.
Kalidas C., Hefter G., Marcus Y. // Chem. Rev. 2000. V. 100. № 3. P. 819.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии