

Журнал физической химии, 2020, T. 94, № 11, стр. 1706-1715
Фрагментация протонированного диметиламиноазобензола при лазерном импульсном фотолизе в криогенной атмосфере разреженного гелия
Ю. А. Михеев a, *, Ю. А. Ершов b
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
b Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 09.01.2020
После доработки 09.01.2020
Принята к публикации 21.01.2020
Аннотация
Проведен анализ данных по составу катионных фрагментов, образующихся при импульсном фотолизе протонированного красителя диметиламиноазобензола в разреженной атмосфере гелия при T ≈ 40 K. Показана несостоятельность трактовки механизма фотолиза на основе традиционного представления этого красителя как мономерного катиона азония. Адекватный механизм процесса получен в рамках новой концепции, согласно которой, основное состояние протонированного диметиламиноазобензола и его замещенных производных – ридимеры с двумя протонированными аминогруппами. Установлено, что фотовозбуждение этих ридимеров в разреженной атмосфере гелия приводит к их депротонированию с образованием электронейтральных мономеров и мономеров с положительными зарядами на sp2-орбиталях; оба типа мономеров реагируют в атмосфере гелия с освободившимися протонами, расщепляясь на разнообразные катионные фрагменты. Приведены формулы строения фрагментов и схемы фрагментации мономеров.
В недавно опубликованных работах [1–5] осуществлен анализ результатов экспериментальных исследований UV-VIS-спектроскопических и структурно-химических свойств аминоазобензольных красителей, накопленных за ≈100 лет. В них показана неадекватность представления, сформулированного на этапе становления органической химии и существующего до сих пор, согласно которым, простые красители на основе аминоазобензола в своем основном состоянии являются мономерами. В реальности их основное состояние – ридберговские димеры (ридимеры). При этом характерная для азокрасителей цветность связана не с их хиноидными мономерами, как полагали до этого более 100 лет, а с принадлежащими ридимерам катионами фениламинильного типа (PhAT).
Результаты работ [1–5] создали условия для реновации накопленных в литературе трактовок экспериментальных данных по фотохимии аминоазобензольных красителей в аспекте их ридимерного строения. В настоящее время установлен факт усиления кислотности протонированных ридимеров аминоазобензола при их фотовозбуждении [6], раскрыты детали фотоники ридимеров 4-аминоазобензола в спиртовой среде [7, 8], метилоранжа в водной среде [9], 4-N,N-диэтиламиноазобензола в изопропаноле [10], а также мономеров протонированного азобензола в криогенной атмосфере гелия [11].
В настоящей работе проанализированы данные [12] о фотофрагментации ридимеров протонированного 4-N,N-диметиламиноазобензола ((ABN+R2H)2, R = CH3) в криогенной атмосфере гелия. Свои результаты авторы [12] рассматривают как подтверждение ключевой роли катионов азония в предложенном ими механизме фотофрагментации протонированного транс-азобензола (t-ABH+). По-мнению авторов [12], молекулярное строение обоих исходных соединений в виде катионов азония подтверждается их ab initio расчетами по современным компьютерным программам. Между тем, из [1–11] следует, что осуществленные в [12] расчеты не позволили раскрыть реального наличия и ключевой роли катионов PhAT в превращениях t-ABH+.
Ключевая роль катионов PhAT в газофазной фотофрагментации t-ABH+ получила объяснение в [11]. Цель настоящей работы – раскрыть механизм фотофрагментации протонированного 4‑N,N-диметиламиноазобензола на основании данных [12] с учетом его ридимерного строения (ABN+R2H)2 и ключевой роли в нем катионов PhAT.
Механизм фотофрагментации в трактовке авторов [12]
Авторы [12] демонстрируют актуальность методики криогенной газофазной фотофрагментации ароматических соединений на примерах протонированных транс-азобензола t-ABH+ и 4‑N,N-диметиламиноазобензола (“DAB-Az+”), традиционно характеризуя их в виде катионов азония. В качестве газовой среды используют разреженный гелий, охлажденный до T ≈ 40 K. Установлено [12], что фотовозбуждение t-ABH+ в криогенной атмосфере гелия приводит к образованию катионов фенила (Ph+) и фенилдиазония (C6H5N+≡N, benzenediazonium, BD+). Эти катионы, вместе с диметиламинофенилкатионами, указаны [12] в качестве главных продуктов криогенной фотофрагментации мнимых мономерных катионов “DAB-Az+”. Полученные результаты авторы [12] рассматривают как свидетельство одинаковых механизмов фотофрагментации мономеров t-ABH+ и “DAB-Az+” с разрывами C–N-связей по обе стороны азогрупп катионов азония по схеме 1.

Согласно схеме 1, фотовозбуждение катиона азония в t-ABH+ вызывает перенос электрона с π-орбиталей колец 1, 2 на протонированный атом азота азогруппы. По мнению авторов [12], эти переходы, осуществляемые то с одного, то с другого кольца, приводят к нейтрализации положительного заряда на группе HN+ с последующим гомолитическим разрывом связи H–N, а образующиеся при этом радикалы H• мигрируют к кольцам, вызывая распад мономера на молекулы бензола (B) и катионы фенилдиазония (BD+), частично диссоциирующие на азот и катионы фенила. Считается, что в ходе молекулярных осцилляций фотовозбужденного катиона азония происходят акты переноса π-электронов с колец на пустую π-орбиталь атома N+, созданную присоединенным протоном.
Такие же акты переноса электронов с фенильного и фениленового колец вызывают, по мнению авторов [12], фрагментацию мнимых мономеров “DAB-Az+”, строение которых они представляют в виде катиона азония (схема 2).

Между тем, электронные (e-) формулы t-ABH+ и ридимеров (ABN+R2H)2 включают в себя катионы PhAT. Именно катионы PhAT, а не азония (схема 2), являются хромогенами протонированных молекул AB и ридимеров аминоазобензольных красителей [1–11]. Например, катион PhAT в t-ABH+ имеет e-формулу (схема 3), исключающую актуальность схемы 1. Так, согласно схеме 1, фрагментация должна идти вслед за актами переноса электрона с того или иного фенильного кольца на pz-орбиталь азота, присоединившего протон (NH+). Однако данная pz-орбиталь в основном состоянии t-ABH+ занята двумя электронами (схема 3), и на ней нет места для третьего электрона. Образование $p_{{\text{z}}}^{2}$ связано с тем, что присоединившийся к азоту протон поляризует π-связь азогруппы так, что оба ее π-электрона проводят время преимущественно на pz-орбитали азота группы NH+. Данная особенность катиона PhAT аннулирует акты фотопереноса электронов, указанные на схеме 1 стрелками, и обеспечивает в t-ABH+ акты фотопереноса с участием катионов PhAT [11].

Данные [12] по мнимым мономерным катионам “DAB-Az+”
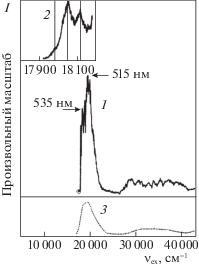
Авторы [12] использовали установку с наносекундным оптическим параметрическим генератором (OPO) с частотой повторения импульсов 10 Гц (спектральное разрешение 8 см–1, шаг сканирования 0.02 нм, энергия в импульсе ≈0.5 мДж). Пучки излучения фокусировали в ловушку с мнимыми катионами “DAB-Az+”, находящимися в разреженной атмосфере охлажденного гелия при низкой температуре ≈40 K. Образование продуктов фотолиза регистрировали с помощью времяпролетного (TOF) масс-спектрометра (спектроскопия фотофрагментационого действия, PFA), представляя их в виде разностного спектра PFA (рис. 1). Кроме того, на основании данных спектроскопии PFA строили UV-VIS-спектры возбуждения фрагментации (PFS) по зависимости суммарного выхода продуктов фрагментации от волнового числа νex, см–1 (рис. 2, спектры 1, 2).
Рис. 1.
Дифференциальный TOF-масс-спектр фотолизованных ридимеров протонированного диметиламиноазобензола (m/q = 226, отрицательный пик). Данные [12]. Пояснения в тексте.

Катионы мнимых мономеров “DAB-Az+” продуцировали в электрораспылительном источнике (electrospray ion source) из раствора c концентрацией 0.1 мМ в смеси вода/метанол = 1 : 1 при небольшом количестве уксусной кислоты. Методика с распылением включает в себя впрыскивание порций раствора через нагретый капилляр в условиях значительного перепада давления в объем с низким давлением ≈1 мБар (100 Па, 0.75 Торр), в котором происходит удаление растворителя. При движении в нагретом капилляре раствор находится под действием высоковольтного напряжения (несколько киловольт), в результате чего образуются сильно заряженные капли [13, 14], распыляющиеся в объеме и теряющие растворитель. В условиях высокого напряжения образующаяся соль (ацетат (ABN+R2H)2) теряет анионы ацетата.
Созданные катионы (ABN+R2H)2 инжектировали в ловушку, охлаждаемую импульсом криогенного гелия. Для создания температуры ≈40 K использовали холодный гелий с давлением 0.1–1.0 мТорр [14]. На охлаждение катионов в ловушке уходило несколько десятков миллисекунд, после чего подавался лазерный импульс для фотофрагментации мнимых катионов “DAB-Az+”. Устройство экспериментальной установки позволяло экстрагировать ионные фрагменты и остатки “DAB-Az+” из ловушки в TOF масс-спектрометр. Разностные спектры PFA представляют собой результаты вычитания спектра исходных “DAB-Az+” (laser off) из спектров катионных продуктов фрагментации (laser on). В качестве примера в [12] приведен разностный спектр PFA, полученный под действием излучения с λ = 275 нм. Этот спектр (рис. 1), позволил авторам [12] заключить, что главными продуктами фотофрагментации мнимых катионов “DAB-Az+” (отношение масса/заряд – m/q = 226) являются фенильные катионы (Ph+, m/q = 77), катионы фенилдиазония (C6H5N$_{2}^{ + }$, benzenediazonium, BD+ c m/q = 105) и диметиламинофенил-катионы (m/q = 120).
Следует отметить, что на спектре PFA (рис. 1) имеются еще несколько сигналов: два достаточно интенсивных пика при m/q между 77 и 105 и m/q между 120 и 226, а также три слабых пика (два при низких и один при высоком m/q). О присутствии этих пиков на спектре PFA (рис. 1) и соответствующих им катионов авторы не упоминают, вероятно, считая достаточным отметить единство механизмов фотофрагментации мнимых “DAB-Az+” и t-ABH+ с разрывом C–N-связей. Между тем, ниже показано, что происхождение этих, “забытых” авторами [12] катионов, получает объяснение в рамках ридимерной концепции. К тому же показатель m/q = 226, приписанный мнимому мономерному катиону “DAB-Az+”, характеризует ридимерный дикатион (ABN+R2H)2 в силу характерного для него соотношения 2m/2q = 226.
На рис. 2 приведены спектры 1, 2 возбуждения фотофрагментации (PFS) мнимого “DAB-Az+”, полученные в [12] на основе спектров PFA в виде зависимости суммы выходов главных продуктов фрагментации (m/q = 77, 105, 120) от волнового числа (νex, см–1). Основанием для использования суммы их выходов стало то, что соотношение интенсивностей сигналов отмеченных фотопродуктов в спектрах PFA является инвариантной функцией от частоты возбуждения νex. Особенность спектра 2 заключается в том, что он характеризует начальный участок спектра 1 (отмечен кружком), перестроенный с высокой степенью усиления. Рассматривая спектры 1, 2 (рис. 2), авторы [12] ограничились замечанием, что их сочетание не соответствует ожидаемой ситуации с 0–0-переходом, а наблюдаемые в VIS-области (ν = 17 000–25 000 см–1) и UV-области (ν = 27 000–41 700 см–1) две полосы выглядят широкими и не позволяют оценить число электронных состояний в спектре.
Дополнительно на рис. 2 авторы [12] привели спектр 3, заимствованный из [15], для демонстрации его практического сходства со спектром 1. В [15] спектр 3 (рис. 2) традиционно приписан мнимым мономерным катионам “DAB-Az+”. (Между тем, согласно [6], авторы [15] имели дело с раствором ридимеров (ABNR2)2 в ацетонитриле с небольшим количеством HСl, достаточным для полного смещения равновесия протонирования к ридимерным дикатионам аммония (ABN+R2H)2).
Обсуждение данных [12] в аспекте ридимерной концепции
Согласно данным [6], использованный в [12] спектр 3 (рис. 2) характеризует равновесие между двумя электронными таутомерами ридимерного дикатиона (ABN+R2H)2, протонированного по обеим аминогруппам (схема 4).
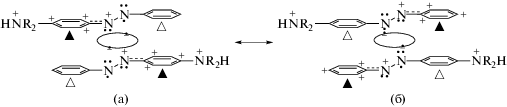
На схеме 4 показано, что e-таутомерный обмен в ридимерах (4a) ↔ (4б) идет путем ультрабыстрой переполяризации мономеров с вовлечением в этот обмен R3s-электронов. Именно такие e-таутомеры генерируют UV–VIS-спектры типа 3 (рис. 2) [1–6]. В частности, отмеченные на схеме 4 светлыми треугольниками смежные пары фенильных и фениленовых колец (не имеющие зарядов) ответственны за UV-полосы при λ ≈ 318 нм (ν ≈ 27 000 см–1). Эти смежные кольца представляют собой бинарные квантово-волновые резонаторы и взаимодействуют друг с другом в возбужденном состоянии по механизму Симпсона [1–6], ответственному за UV-полосу при λ ≈ 318 нм.
Отмеченные черными треугольниками смежные пары фенильных и фениленовых колец (схема 4) ответственны за две налагающиеся друг на друга VIS-полосы дипротонированных ридимеров (ABN+R2H)2 с максимумами при 515 и 535 нм (рис. 2, спектры 1, 3). Указанные VIS-полосы тоже появляются в результате взаимодействия положительно заряженных парных фенильных и фениленовых фрагментов по механизму бинарных резонаторов Симпсона [1–6].
На рис. 2 спектр 2 дан авторами [12] с высокой степенью усиления сигнала (он характеризует начальный участок спектра 1 с νex ≥ 17 900 см–1, отмеченный кружком). При этом преследовали цель установить спектральное единообразие со спектром возбуждения фотофрагментации (PFS) мнимого азоний-катиона t-ABH+ на начальном участке. В случае мнимого азоний-катиона t‑ABH+ высокое усиление сигнала на начальном участке спектра UV-VIS PFS позволило установить наличие низкочастотной колебательной (v) последовательности, которую с помощью квантовых расчетов [12] связали с торсионными движениями диэдрального угла CNNC в S1-состоянии. Однако, структура начального участка на рис. 2 (спектр 2) не получила соответствующего объяснения.
В [11] появление низкочастотной колебательной последовательности на начальном участке спектра PFS мнимого азоний-катиона t-ABH+ связали с суперпозицией эффектов резонансного рамановского (RR) рассеяния и e-фотовозбуждения, результат которой четко проявляется на самом начальном низкочастотном участке полосы e-перехода. Этого следует ожидать и в случае со спектром фотофрагментации (PFS) мнимого азоний-катиона“DAB-Az+”.
Природа низкоэнергетической вибронной структуры в PFS-спектре
Рамановское рассеяние и e-фотовозбуждение связаны с внутримолекулярными колебательными (v) процессами и, активируя нормальные колебания, способны индуцировать обратную связь между собой. Эта связь значительно усиливается в режиме резонансного рамановского (RR-) рассеяния, когда частота света νex, возбуждающего рассеяние, налагается на спектральную полосу поглощения [16].
Если в молекуле связь между нормальными колебаниями невелика, и при e-переходе возбуждается лишь определенная группа атомов, то избыток v-энергии будет сосредоточен в этой группе. При этом сама молекула может считаться простой в спектроскопическом отношении [17, 18]. С усложнением молекулы и увеличением запаса ее колебательной энергии (за счет повышения температуры, увеличения νex) возрастает связь нормальных колебаний между собой и с электронным состоянием. В таком случае растет вероятность внутримолекулярного перераспределения v-энергии, и в v-процесс вовлекается значительная часть ансамбля или весь ансамбль нормальных колебаний молекулы. В предельном случае в молекуле достигается термическое равновесие, характеризуемое колебательной температурой, и молекула считается спектроскопически сложной [17, 18].
В полусложных молекулах перераспределение энергии e-возбуждения не доходит до термического равновесия, и их e-спектры имеют явно выраженную колебательную структуру. При этом избыток v-энергии, не успевая распределиться по всем колебательным степеням свободы, сохраняется на большей или меньшей их части до тех пор, пока не произойдет акт физической или химической трансформации запасенной e,v-энергии, например, флуоресценция или диссоциация (фрагментация). Наличие на начальном участке спектра 2 (рис. 2) крайне слабой по интенсивности вибронной структуры позволяет отнести мнимые азоний-катионы “DAB-Az+” (реально, дипротонированные ридимеры (ABN+R2H)2), а также t-ABH+ [11], к спектроскопически полусложным объектам, для которых возможны перечисленные ниже ситуации.
С одной стороны, поглощаемое излучение вызывает, e-переход (S0 → S1, время акта ∼1 фс) и последующую фрагментацию ридимера (ABN+R2H)2, с другой стороны, оно генерирует акты RR-рассеяния с появлением v-возбужденных состояний $S_{0}^{{\text{v}}}$ (время актов ∼1 фс). В общем случае энергия v-возбужденных ридимеров $S_{0}^{{\text{v}}}$, появляющихся под действием актов возбуждения за время лазерного импульса с частотой νex, может расходоваться в параллельных процессах: 1) генерации антистоксовых полос [16], 2) перераспределения по колебательным степеням свободы ридимера, причем с преимущественной аккумуляцией на низкоэнергетических v-уровнях, 3) в актах повторного фотовозбуждения ($S_{0}^{{\text{v}}}$ → $S_{1}^{{\text{v}}}$) с последующей фрагментацией.
Наличие пиков на спектре 2 (рис. 2) свидетельствует о том, что фрагментационное e-энергетическое сканирование лазерными импульсами (вдоль оси νex) позволяет обнаружить достаточное интенсивное перераспределение энергии на низкорасположенный v-уровень. v-Энергия этого уровня суммируется в ходе лазерного импульса с энергией повторного e-возбуждения ($S_{0}^{{\text{v}}}$ → $S_{1}^{{\text{v}}}$) при том, что вероятность актов фрагментации становится превалирующей над конкурентными актами потери v-энергии. В данном случае импульсы с νex исполняют роль сканирующего спектроскопического зонда в отношении тех низкоэнергетических колебаний в ридимере, которые инициируют акты фрагментации (схема “pump + probe ← puls’’).
Выше отмечалось, что PFS-спектры 1, 2 (рис. 2) получены [12] по зависимости суммы выходов главных продуктов фрагментации (m/q = 77, 105, 120) от волнового числа νex на основании того, что расположение сигналов этих катионов на спектрах PFA не зависит от νex. Наличие такой инвариантности (она отсутствует у PFA-спектров t‑ABH+ [11]) свидетельствует, что разные по природе катионные фрагменты образуются вследствие единого для них акта фотоинициирования. Такое единство характерно уже для самого начального участка VIS-полосы (рис. 2, спектр 2), и оно отражает суперпозицию энергии, вносимой с νex, и энергии низкоэнергетического v-движения. Оценка энергии этого v-движения, сделанная по разности значений максимумов νex соседних полос на рис. 2, спектр 2 (обозначены вертикальными линиями), дает величину Δν ≈ 60 см–1.
Следует отметить, что частота Δν ≈ 60 см–1 находится рядом с низкочастотной полосой Δν = 47 ± ± 12 см–1, обнаруженной методом резонансной импульсной стимулированной рамановской спектроскопии (RISRS) для растворенного в диметилсульфоксиде непротонированного 4'-нитро-4-диметиламиноазобензола, традиционно рассматриваемого как мономерное соединение [19]. Между тем, давно установлено, что данный краситель находится в виде димера в парообразном состоянии [20], а недавно показано, что по своей природе он является ридимером и существует также в жидкой среде [3]. Этот ридимер протонируется по аминогруппам и сохраняется в виде ридимера при средней кислотности раствора [3].
Что касается низкочастотной полосы (Δν = = 47 ± 12 см–1) 4'-нитро-4-диметиламиноазобензола, то квантовые расчеты позволили авторам [19] отнести ее к внутриплоскостным (in plane) размашистым (sweeping) колебаниям фенильных колец вместе с их заместителями. Обнаруженная в той же работе [19] значительно более слабая и более низкочастотная полоса Δν = 14 ± 10 см–1 была отнесена к крутильным (torsional) движениям фенильных колец вокруг центральных C–N-связей. Следует отметить, что соотношение интенсивностей отмеченных полос естественным образом согласуется с ридимерным строением 4'‑нитро-4-диметиламиноазобензола. Действительно, фиксированное ридберговской связью соседство двух мономеров в параллельных плоскостях оказывает значительные препятствия для крутильных движений фенильных колец, оставляя свободу для широких движений в плоскостях мономеров.
На основании изложенного, ридимер 4'-нитро-4-диметиламиноазобензола можно рассматривать как референтное соединение для ридимера (ABN+R2H)2, чьи низкоэнергетические v-колебания с Δν ≈ 60 см–1 тоже должны отражать внутриплоскостные колебания в состоянии $S_{0}^{{\text{v}}}$. При этом суммирование энергии таких v-колебаний с энергией сканирующей частоты νex, создает состояние $S_{1}^{{\text{v}}}$, которое становится причиной распада ридимера (ABN+R2H)2 на катионные фрагменты и появления соответствующей вибронной структуры в фотофрагментационном спектре 2 (рис. 2). Таким образом, размашистые внутриплоскостные колебания фенильных колец способствуют актам отщепления протонов от ридимеров (ABN+R2H)2. Отметим, что фотодепротонирование ридимеров (ABN+R2H)2 было установлено ранее для его растворов в ацетонитриле, содержащем HCl [3].
Фотохимические реакции ридимеров (ABN+R2H)2
Согласно [3], действие VIS-излучения с λex = = 514 нм (νex = 19 455 см–1) на (ABN+R2H)2 в среде ацетонитрила приводит к обратимой реакции депротонирования, в которой освобожденный протон сольватируется полярными молекулами растворителя, и один из мономеров ридимера получает хиноидное строение в соответствии со схемой 5.

Появление хиноидных структур (5б) регистрируется по возникновению интенсивной RRS-полосы с ν =1623 см–1 [3]. За поглощение света с λex = 514 нм ответственны бинарные хромогены из смежных катионов PhAT (отмечены черными треугольниками в схемах 4, 5). Излучение с λex = = 351 нм (νex = 28 490 см–1), поглощаемое бинарными хромогенами из смежных электронейтральных фенильных и фениленовых колец (отмечены светлыми треугольниками в схемах 4, 5), вызывает разрушение ридимеров (ABN+R2H)2, причем настолько сильное, что хиноидная RRS-полоса с ν = = 1623 см–1 практически аннулируется [3]. Механизм разрушения можно прояснить с учетом данных по фрагментации (ABN+R2H)2 [12].
Механизм фотофрагментации (ABN+R2H)2
Облучение раствора ридимеров (ABN+R2H)2 в ацетонитриле [15] светом с λex = 514 нм обеспечивает состояние равновесия по схеме 5 вследствие наличия протонов ${\text{H}}_{{{\text{solv}}}}^{ + }$ от диссоциированной соляной кислоты. Для жидкой среды [15] характерно не только наличие ${\text{H}}_{{{\text{solv}}}}^{ + }$, но и жидкофазного клеточного эффекта [21], который тоже создает определенные препятствия для расщепления ридимеров (5б) на мономеры. Атмосфера разреженного гелия [12] не содержит протонов, а появляющиеся при фотодиссоциации ридимеров (ABN+R2H)2 протоны не имеют возможности сольватироваться и способны лишь образовывать неустойчивые комплексы с атомами He, которые становятся переносчиками H+.
Отсутствие ${\text{H}}_{{{\text{solv}}}}^{ + }$ и клеточного эффекта обеспечивает: 1) необратимость образования ридимеров (5б); 2) диссоциацию ридимеров (5б) на мономеры и распад мономеров на разнообразные по значениям m/q катионные фрагменты. Этому способствует наличие аммониевой группы N+R2H у второго мономера (схема 5б). В ней присоединенный протон индуцирует распределение положительного заряда по четырем связям азота аминогруппы [22], увеличивая степень делокализации заряда. Эта аммониевая группа попадает в зону перекрывания вторых сферических пучностей 3s-орбиталей обеих азогрупп, участвующих в формировании ридберговской межмономерной связи. Возникает возможность для рекомбинации положительного заряда группы N+R2H и электрона ридберговской связи с отщеплением атома H.
Изложенное выше можно представить в виде схемы 6.
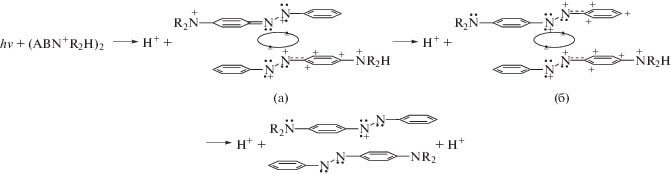
На схеме 6 показано, что после первичного акта депротонирования ридимерный таутомер (6а) пререходит в (6б) путем переполяризации азогруппы в верхнем таутомере вследствие обмена электронов между sp2-орбиталями при участии электронов ридберговской связи по схеме ${\text{R}}_{{{\text{3s}}}}^{{ \bullet \bullet }}$ + + ${\text{N}}_{{ \bullet \bullet }}^{{}}$–${\text{N}}_{{ + \bullet }}^{{ \bullet \bullet }}$ ↔ ${\text{R}}_{{{\text{3s}}}}^{{ \bullet + }}$ + ${\text{N}}_{{ \bullet \bullet }}^{{}}$–${\text{N}}_{{ \bullet \bullet }}^{{ \bullet \bullet }}$ ↔ ${\text{R}}_{{{\text{3s}}}}^{{ \bullet \bullet }}$ + ${\text{N}}_{{ \bullet + }}^{{ \bullet \bullet }}$–${\text{N}}_{{ \bullet \bullet }}^{ + }$. Связанное с этим появление на мономерах одинаковых зарядов по одну сторону ридимера (6б) приводит к их отталкиванию и способствует диссоциации таким образом, что один из электронов разрывающейся межмономерной связи рекомбинирует с положительным зарядом группы N+R2H в (6б), вызывая потерю протона. Второй электрон от разорванной ридберговской связи рекомбинирует с положительным зарядом sp2-орбитали того или другого мономера, реализуя итоговую картину в соответствии со схемой (6).
Таким образом, отличие процесса фотовозбуждения ридимеров в атмосфере гелия (схема 6) от жидкофазного процесса с λex = 514 нм (схема 5) заключается в появлении двух протонов и мономеров, из которых один несет заряд на sp2-орбитали азогруппы, а другой является электронейтральным. В связи с этим необходимо рассмотреть две неодинаковые цепочки фрагментации с участием положительно заряженных и электронейтральных мономеров.
Схема фрагментации положительно заряженных мономеров
Положительно заряженному мономеру соответствует обратимое равновесие двух e-таутомеров, связанных с переходом катионов PhAT между кольцами:
Учитывая это, естественно полагать, что протон будет атаковать незаряженные фенильное и фениленовое кольца, индуцируя приведенные ниже реакции:

Эти реакции объясняют образование идентифицированных в [12] фенилкатионов Ph+ (m/q = = 77) и диметиламинофенилкатионов R2NPh+ (m/q = 120), а также неидентифицированных в [12] катионов азота N2+ (m/q = 28) и диметиламмонийкатиона R2N+ (m/q = 44).
Схема фрагментации электронейтральных мономеров
Атака протона на фенильное кольцо электронейтрального мономера приводит к неидентифицированному в [12] (диметиламино)фенилдиазониевому катиону (R2NBD+, m/q = 148) и идентифицированному диметиламинофенилкатиону (R2NPh+, m/q = 120):
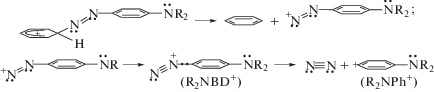
При атаке протона на фениленовое кольцо возможны два варианта:
и ведущие к образованию BD+ (m/q = 105), фенилкатиона Ph+ (m/q = 77) и диметиламмонийкатиона R2N+ (m/q = 44).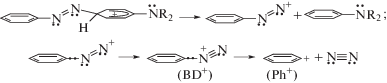
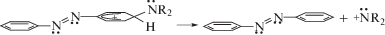
Предсказываемое приведенными выше схемами образование фрагментных катионов из ридимеров (ABN+R2H)2 мы верифицировали путем построения графика зависимости соответствующих им значений m/q от моментов времени (t, мкс) их регистрации на спектрах TOF (рис. 1). Этот график приведен на рис. 3. Из рис. 3 видно, что все экспериментальные точки работы [12] вполне удовлетворительно соответствуют единой прямой m/q = k(t + 14) (k – константа), как для идентифицированных в [12] катионных фрагментов (светлые квадраты), так и для идентифицированных нами катионов (черные кружки). Данный факт однозначно свидетельствует об адекватности механизма фотофрагментации именно ридимеров (ABN+R2H)2, а не мономерных катионов азония “DAB-Az+”.
Рис. 3.
Обобщенный график выхода катионных фрагментов, построенный по TOF-спектру (рис. 1). Пояснения в тексте.

Следует отметить, что на рис. 3 указаны еще катионы с m/q = 93 и 155, отсутствующие в приведенных выше реакционных схемах. Их наличие свидетельствует о том, что в реакционном объеме идет с невысокими выходами присоединение осколочных катионов к молекулам образующегося бензола (B). Так, сигнал m/q = 93, может принадлежать σ-комплексам (СH3B+) молекул бензола с метилкатионами, образующимся с невысоким выходом в первичных актах параллельно актам депротонирования. Сигнал m/q = 155 может принадлежать σ-комплексам фенилкатионов и молекул бензола – PhB+. Кроме того, на TOF-спектре (рис. 1) рядом с сигналом m/q = 77 заметен весьма слабый сигнал, соответствующий m/q = 79, что может указывать на образование σ‑комплексов HB+.
Согласно данным рис. 2, установленные нами катионные фрагменты образуются не только в VIS-интервале (18 000–25 000 см–1), но и в UV-интервале излучения (25 000–40 200 см–1). Это можно связать, во-первых, с UV-возбуждением в полосе поглощения бинарных хромофоров (состоящих из смежных электронейтральных фенильных и фениленовых колец – отмечены светлыми треугольниками в схемах 4, 5, область поглощения при λ ≈ 318 нм (ν ≈ 27 000 см–1)) и, во-вторых, с UV-поглощением одиночных фенильных и фениленовых групп в той спектральной области (λ < 318 нм (ν > 27 000 см–1)), где теряет силу условие квантово-волнового резонанса по механизму Симпсона. Сам факт UV-фрагментации свидетельствует о наличии внутриридимерных актов передачи энергии возбуждения на те ридимерные e-конфигурации, которые являются инициаторами фрагментации (схемы 5, 6).
Реализация таких же актов переноса энергии позволяет объяснить установленный в [6] факт преимущественного разрушения ридимеров (ABN+R2H)2 в среде ацетонитрила при действии UV-излучения с λex = 351 нм (νex = 28 490 см–1).
Таким образом, наш анализ данных [12] в аспекте ридимерной концепции позволяет заключить, что 1) использованная в [12] модель мономерного азоний-катиона протонированного диметиламиноазобензола несостоятельна для объяснения его фотофрагментации так же, как и использованные для него расчеты ab initio, 2) построение адекватного механизма данного процесса возможно только на основе ридимерного строения исходного красителя, 3) поглощаемое UV-VIS-излучение индуцирует расщепление ридимеров (ABN+R2H)2 на мономеры с образованием протонов, которые в реакциях с мономерами генерируют широкий набор фрагментов из катионов, свободных радикалов и валентно насыщенных молекул.
Список литературы
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2017. Т. 91. № 4. С. 672.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2017. Т. 91. № 10. С. 1683.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 2. С. 267.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 8. С. 1251.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 10. С. 1552.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 2. С. 313.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 6. С. 946.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 7. С. 1111.
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 11. С. 1746–1755.
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94. № 1. С. 143.
Михеев Ю.А., Ершов Ю.А. // Там же. (В печати).
Féraud G., Dedonder-Lardeux C., Jouvet C., Marceca E. // J. Phys. Chem. A. 2016. V. 120. № . P. 3897.
Andersen J.U., Hvelplund P., Nielsen S.B., Tomita Sh. et al. // Rev. Sci. Instruments. 2002. V. 73. № 3. P. 1284.
Wang Xue-Bin, Wang Lai-Sheng // Rev. Sci. Instruments. 2008. V. 79. № 7. P. 073108.
Matazo D.R.C., Ando R.A., Borin A.C., Santos P.S. // J. Phys. Chem. A. 2008. V. 112. № 19. P. 4437.
Fujino T., Tahara T. // J. Phys. Chem. A. 2000. V. 104. № 18. P. 4203.
Борисевич Н.А. Внутримолекулярное перераспределение колебательной энергии в взбужденном состоянии. В сб. “Элементарные фотопроцессы в молекулах”. Отв. ред. Б.С. Непорент. М.-Л.: Наука, 1966. С. 55.
Непорент Б.С. “Природа сплошных вибронных спектров сложных молекул”. В сб. “Молекулярная фотоника”. Отв. ред. А.А. Красновский, Б.С. Непорент. Л.: Наука, 1970. С. 18.
Hoffman D.P., Ellis S.R., Mathies R.A. // J. Phys. Chem. A. 2013. V. 117. № 45. P. 11472.
Green H.S., Jones F., Syz M., Zollinger Hch. // Helvetica Chimica Acta. 1965. V. 48. № 45. P. 389.
Колдин Е. Быстрые реакции в растворе. М.: Мир, 1966. С. 282–284.
Темникова Т.И. Курс теоретических основ органической химии. Л.: Гос. науч.-тех. изд-во хим. лит., 1962. С. 64.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии