

Журнал физической химии, 2020, T. 94, № 4, стр. 595-601
Влияние амино- и фосфорсодержащих алкиленовых заместителей на комплексообразующую способность фосфорсодержащих катионитов по отношению к катионам индия и галлия
Б. К. Радионов a, Ю. А. Лейкин b, А. Л. Смирнов a, И. А. Свирский a, *
a Уральский Федеральный университет им. Б.Н. Ельцина
Екатеринбург, Россия
b Российский химико-технологический университет им. Д.И. Менделеева
Москва, Россия
* E-mail: svirskill.171993@gmail.com
Поступила в редакцию 22.05.2019
После доработки 22.05.2019
Принята к публикации 18.06.2019
Аннотация
Варьированием природы и взаимным расположением дополнительных основных и кислотных групп фосфиновокислотных макропористых ионитов осуществлено регулирование их координационных свойств при сорбции катионов индия и галлия. Определены закономерности направленного изменения кислотной и электронодонорной способности полидентатных фрагментов на сорбционно-избирательные характеристики ионитов при исключении влияния матричной селективности. Показано, как заместители в виде протоноакцепторных анионогенных и фосфоновокислотных групп за счет увеличения ионно-координационной силы фосфорильной системы влияют на сорбируемость рассеянных элементов. Установлено, что благоприятное пространственное строение дифосфоновых катионитов и эффект дополнительного цикла повышают степень поглощения, особенно, галлия в сернокислых растворах; модификация ионитов путем замены заместителей в фосфиновом центре служит плодотворной базой для создания ряда новых хелатогенных сорбентов.
В предыдущей работе [1] продемонстрирована возможность качественного изменения комплексообразующих свойств и сорбционных показателей по рассеянным элементам фосфорсодержащих катионитов путем замены одной кислотной ОН-группы, практически не участвующей в сорбционном процессе из-за ее слабокислотных свойств и высокого значения кислотности среды, рядом алкильных заместителей, повышающих активность координационного центра. Сорбционная способность фосфиновокислотных катионитов с алкил- и алкоксил-радикалами линейно зависит от кислотной диссоциации ОН-групп (pKа), констант Кабачника ($\sigma _{{\text{ф}}}^{*}$) и электроотрицательности заместителей. Этими типами радикалов не ограничивается круг перспективных заместителей, достойны внимания и такие группировки, которые не только смещают электронное облако фосфинового центра в сторону фосфорильного кислорода, но и обладают собственной координационной способностью [2, 3, гл. 2].
Такими свойствами характеризуются акцепторные заместители с ионогенными амино- и фосфониевыми алкилами, включая целостную фосфоновую группу. В качестве кислотных заместителей предпочтение, безусловно, отдано фосфоновым кислотам, увеличивающим функциональную дентатность анкерной Р–R–Р-связки. Конкретными объектами изучения послужили иониты с заместителями разной природы и местом их расположения, когда дополнительные алифатические радикалы связаны с главным Р-центром посредством мостиковой цепи аминным или фосфинным концом. Фосфониевые группы –Р+RХ– по основной силе близки к аммониевым основаниям. По существу, такие N- или P+-замещенные фосфиновые иониты с амбивалентными функциональными группировками кислотного и основного характера имеют все признаки амфотерных ионитов.
В этой связи следует напомнить, что отличительной чертой сорбционного поведения рассеянных элементов на амфолитах с функциональными группами α-аминометилфосфоновой кислоты и катионитах с фосфоновокислотными группами является различная зависимость сорбируемости от уровня кислотности внешнего раствора, которая у первых сохраняется с высокими емкостными показателями в сильно концентрированных кислотосодержащих средах [4, 5]. Однако характер взаимодействия катионов индия и галлия с аминофосфоновыми группами амфолита подобен механизму их координации с фосфорсодержащими катионитами. И в том, и в другом случаях наблюдается образование хелатной структуры с кислородами фосфоновой группы, но при этом аминогруппы полиамфолитов прямого участия в реакции комплексообразования с ионами металлов не принимают [4, 6, 7].
Изучение закономерностей сорбционного поведения рассеянных элементов в зависимости от природы и функциональной силы реакционных центров проводили на амфотерных ионитах с фрагментами α-, β- и γ-аминоалкиленфосфиновых и α-, γ-фосфонийалкиленфосфиновых кислот. Достоинство этого класса амфолитов – не только четкое расположение кислотного и основного центров, расстояние между которыми создается изначально при синтезе ионита, но и возможность устранения различий, привносимых полимерным остовом. Если на амфолитах с аминоалкиленфосфоновыми кислотами, полученных полимераналогичными превращениями аминополимеров с присоединением ионогенной группы к сетчатой цепи аминным концом [8–10], влияние слабоосновной группы на комплексообразование неразрывно связано с природой пространственной сетки, которые, несомненно, взаимно накладываются, то в фосфиновокислотных фосфоний- и азотсодержащих ионитах, где на полимерном носителе закреплен не азотный, а фосфорный центр, удается независимо оценить влияние аполярной группировки, используя в качестве заместителя при атоме фосфора кислотного узла различные по своей природе разнооснóвные ответвления, и все это на однотипном полистирольном каркасе.
ЭКСПЕРИМЕНТАЛЬАЯ ЧАСТЬ
В концептуальном и методическом плане работа выполнена аналогично предшествующей [1]. В качестве объектов исследования использовали ядернозамещенные фосфиновые иониты, являющиеся продуктами полимераналогичных превращений на идентичных трехмерных макропористых сополимерах (20% ДВБ и 80% изооктан), но с заместителями кислого и основного характера – амфолиты с группами α, β, γ-амино(фосфоний)алкиленфосфиновых кислот и фосфиново-фосфоновые катиониты с фенил-Р-оксиэтилидендифосфоновой кислотой в α, β, γ-положении к фосфиновому центру [2, 3, гл. 2.4]:
• АФ-11, АФ-13 и АФ-14 – ряд α-аминоалкиленфосфиновокислотных амфолитов с различной основностью аминогрупп (первичные –NH2, вторичные –NHC2H4OH и третичные –N(C2H5)2 соответственно) в замещенной фосфиновокислотной группе. Иониты Аr-Р(О)(ОН)СН2NR1R для АФ-11 R = R1 = H, для АФ-13 R = R1 = C2H5, для АФ-14 R = H, R1 = C2H4OH получали полимераналогичными превращениями катионитов с группами α-окси(α-хлор)-метиленфосфиновой кислот (СФМ-1) с различными аминопроизводными по реакции Кабачника–Филдса;
• АФФ-1 – ${\text{Аr}}{\kern 1pt} - {\kern 1pt} {\text{Р}}\left( {\text{О}} \right)\left( {{\text{ОН}}} \right){\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{P(}}{{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{7}}}})_{3}^{ + }$ α‑фосфонийметиленфосфиновокислотный амфолит, синтезировали подобно АФ-11, АФ-13, АФ-14 обработкой полистирол-α-хлорметиленфосфиновых кислот фосфином P(C3H7)3 после замещения ОН-групп в катионите СФМ-1 на хлор под действием соляной кислоты;
• АФ-31, АФ-33, АФ-34, АФФ-3 – серия γ‑амино(фосфоний)алкиленфосфиновокислотных амфолитов с межцентровым фрагментом –СН2–СН(ОН)–СН2– и различными основными группами в соответствующем гомологическом ряду. Эти амфолиты получали Р- и О-алкилированием катионитов СФ-3 и СФ-5 эпихлоргидрином с последующим присоединением аминов или фосфина по хлоргидридной и эпоксидной группе;
• АФ-21 и АФ-30 – β- и γ-аминоалкиленфосфиновокислотные амфолиты с первичными аминогруппами
в алкильном заместителе и соответственно с двойными и тройными метиленовыми мостиками
в межцентровом фрагменте. Их изготовляли с помощью Р-алкилирования групп катионита
СФ-3 этиленамином  и аллиламином (СН2 = СН–СН2–NH2–);
и аллиламином (СН2 = СН–СН2–NH2–);
• СДФ-12 – дифосфоновый катионит Аr–Р(О)(ОН)–C(CH3)(OH)–P(O)(OH)2, содержащий помимо ядернозамещенных фосфиновых групп дополнительные фосфоновые группы, соединенные с фосфином α-алкиленовым мостиком. Такой катионит с группами α-оксиметилендифосфоновой кислоты получали в результате обработки поливинилдихлорфенилфосфина уксусным ангидридом (СН3СО)2О и соединениями трехкоординированного фосфора (PCl3, H3PO3);
• СДФ-20 – дифосфоновый катионит Аr–Р(О)(ОН)–(CH2)2–P(O)(OH)2, в котором фосфоновая группа соединена с ядернозамещенной фосфиновым центром этиленовым мостиком. Катионит с группами получали путем алкилирования фосфиновых групп катионита СФ-3 эфирами винилфосфоновой кислоты С2Н3Р(О)(ОR)2, завершаемого кислотным гидролизом;
• СДФ-32, СДФ-32А – катиониты на базе СФ-3 с группами фенил-Р-β-оксипропилен-α,γ-дифосфоновой и фенил-Р-α-оксипропилен-α,γ-дифосфоновой кислот, которые вводили путем реакции с γ-фосфонатпропионовым альдегидом;
• СДФ-16 катионит с группами фенил-Р-α-оксиметилен-α-фенил-α, α'-дифосфоновой кислоты – образование в структуре α-кетофосфиновокислой группировки Аr–Р(О)(ОН)–C(C6H5) (OH)–P(O)(OH)2 происходит при взаимодействии низкомолекулярного кетофосфоната С6Н5С(О)Р(О)(С2Н5)2 с полистиролфосфонистой кислотой (СФ-3).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Анализ полученных результатов (табл. 1) позволяет констатировать, что введение основных групп ≡N и ≡Р+ увеличивает степень сорбционного сродства ионов индия и галлия к фосфоразот- и фосфорсодержащим амфолитам от γ- к α-производным, коррелируя с повышением трансмиссионного эффекта, передаваемого через межцентровые Р–N- и Р–Р-сегменты. Относительно слабовыраженное влияние (по сравнению с числом членов в межцентровом фрагменте) оказывает тип азотного заместителя –NH2 и –NR2 при фосфоре. Особо сильное воздействие обнаруживают аминоэтанольные группы –NH(CH2)2OH, что связано, вероятнее всего, не с основностью азота, а с присутствием оксисоставляющей. В этой связи уместно отметить амфотерные иониты с группами γ-аминоалкиленфосфиновых кислот, содержащие дополнительную в β-положении гидроксильную группу. Сравнение сорбционных показателей амфолитов с группами γ-амино-β-оксипропилфосфиновой и γ-аминопропил-фосфиновой кислот показывает преимущество первых. В целом, как положительное воздействие следует оценить и присутствие в составе массивного заместителя спиртовых звеньев. Причину этого явления следует искать в облегчении процесса проникновения сорбата в полимерную фазу. Дело в том, что гидратационное воздействие основной группы, находящейся в непосредственной близости к кислотной, несколько ослаблено в сравнении с изолированной ввиду формирования бетаиновой структуры. А резинат внутренней соли вследствие взаимного компенсирования зарядов полярных групп характеризуется изоэлектрическим состоянием:
и пониженной сольватирующей способностью трехмерного полимера с вытекающими отсюда диффузионными затруднениями при сорбции.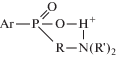
Таблица 1.
Сорбируемость ионов индия и галлия (мг-экв/г) фосфиновокислотными ионитами с основными и кислыми заместителями и на основе макропористого сополимера стирола и ДВБ (Аr – арильный фрагмент стирол дивинилбензольной матрицы)
| Марка ионита | Функциональные группы | 0.1 н. H2SO4 | 0.5 н. H2SO4 | ||
|---|---|---|---|---|---|
| In(III) | Ga(III) | In(III) | Ga(III) | ||
| Амфолиты с группами α-(β-,γ-)амино- и α-(γ-)галогенидфосфонийалканфосфиновых кислот | |||||
| АФ-11 | Аr–Р(О)(ОН)СН2–NH2 | 3.09 | 2.72 | 2.36 | 1.53 |
| АФ-21 | Аr–Р(О)(ОН)CH2CH2–NH2 | 2.89 | 2.07 | 2.04 | 0.98 |
| АФ-31 | Аr–Р(О)(ОН)CH2CH(OH)CH2–NH2 | 2.10 | 1.27 | 1.44 | 0.65 |
| АФ-13 | Аr–Р(О)(ОН)CH2–N(C2H5)2 | 2.76 | 2.34 | 2.14 | 1.26 |
| АФ-33 | Аr–Р(О)(ОН)CH2CH(OH)CH2–N(C2H5)2 | 1.91 | 1.18 | 1.28 | 0.78 |
| АФ-14 | Аr–Р(О)(ОН)CH2–NH–C2H4OH | 2.95 | 2.66 | 2.31 | 1.45 |
| АФ-34 | Аr–Р(О)(ОН)CH2CH(OH)CH2–NH–C2H4OH | 2.21 | 1.36 | 1.54 | 0.69 |
| АФ-30 | Аr–Р(О)(ОН)(CH2)3–NH2 | 1.81 | 1.08 | 1.11 | 0.59 |
| АФФ-1 | Аr–Р(О)(ОН)CH2–P(C3H7)$_{3}^{ + }$ | 1.25 | 0.71 | 0.94 | 0.28 |
| АФФ-3 | Аr–Р(О)(ОН)CH2CH(OH)CH2–P(C3H7)$_{3}^{ + }$ | 2.32 | 1.53 | 1.68 | 0.62 |
| Катиониты с группами 1-окси-1,1-алкандифосфоновых кислот | |||||
| СДФ-12 | Аr–Р(О)(ОН)–C(CH3)(OH)–P(O)(OH)2 | 3.24 | 3.44 | 3.00 | 2.96 |
| СДФ-20 | Аr–Р(О)(ОН)–(CH2)2–P(O)(OH)2 | 2.37 | 1.92 | 1.86 | 1.15 |
| СДФ-32 | Аr–Р(О)(ОН)–CH2–CH(OH)–CH2–P(O)(OH)2 | 3.08 | 2.63 | 2.62 | 2.04 |
| СДФ-32А | Аr–Р(О)(ОН)–CH(OH)–CH2–CH2–P(O)(OH)2 | 3.11 | 2.08 | 2.67 | 2.41 |
| СДФ-12А | Аr–Р(О)(ОН)–C(CH2OH)(OH)–P(O)(OH)2 | 3.18 | 3.76 | 2.87 | 2.59 |
| СДФ-16 | Аr–Р(О)(ОН)–C(C6H5)(OH)–P(O)(OH)2 | 3.10 | 2.72 | 2.65 | 2.07 |
Корреляционные зависимости, связывающие изменение показателей сорбции ионов индия и галлия с электронодонорным эффектом оксисодержащих заместителей и кажущимися константами кислотной диссоциации фосфиновокислотных групп, свидетельствуют (рис. 1) о нивелировании влияния кислотных свойств на степень сорбции индия, а в случае галлия монотонный спад сорбируемости идет по мере снижения способности к кислотной диссоциации замещенных фосфинов. Такое различие обусловлено тем, что ионы индия, по-видимому, более склонны к координационному взаимодействию с фосфорильным кислородом, и за счет выигрыша энергии при хелатообразовании с фосфиновым центром они с легкостью вытесняют протон из кислотного остатка. Более того, с ростом кислотности внешнего раствора, несмотря на снижение степени диссоциации кислотных составляющих фосфиновой системы, просматривается тенденция увеличения сорбируемости индия (III) ионитами с заместителями при атоме фосфора нейтрального и основного характера, которые реализуют возможность смещать электронную плотность на фосфорильный кислород. Формирование хелатных циклов менее характерно для галлия, и поэтому усиление протоноакцепторных свойств слабокислотных групп негативным образом влияет на протекание обменного процесса.
Рис. 1.
Зависимости удельной сорбируемости индия и галлия от кислотных свойств (pKα1,) и констант заместителей ($\sigma _{{{\text{расч}}}}^{*}$) ионитов с группами замещенных фосфиновых кислот: 1– In (0.1 N H2SO4), 2 – In (0.5 N H2SO4), 3 – Ga (0.1 N H2SO4), 4 – Ga (0.5 N H2SO4).

Наличие четвертичной фосфониевой группы в том же положении к фосфорному центру, что и аминогруппа, не вносит радикальных перемен в сорбционное поведение рассеянных элементов в сравнении с первичными, вторичными или третичными аминами. Переход к ионитам с меньшим количеством звеньев между биполярными Р– R – Р+-группами сопровождается увеличением показателей сорбции. Коэффициенты распределения ионов индия и галлия закономерно уменьшаются по мере увеличения числа метильных звеньев, разделяющих N- и P-ионогенные группы и фосфиновый центр, в связи с затуханием возможности электронной передачи.
Аналогичные полиамфолиты с четвертичными аммониевыми группами не изучены в связи с невозможностью получения амфолита с высоким содержанием кватернизованного азота из-за малой конверсии реакции аминирования. Также не является предметом рассмотрения и полный ряд β-производных амино-(фосфоний)алкиленфосфиновых амфолитов ввиду характерно аномальной легкости β-элиминирования.
Отличительная черта ионитов с амино- и фосфониевыми группами в алкильном заместителе фосфинового центра – более высокая избирательность сорбции ионов индия и галлия, чем у фосфиновых катионитов с алкильными, алкоксильными или гидроксилсодержащими заместителями при общем сохранении порядка сорбции рассеянных и цветных металлов In3+ > Ga3+ > > Cu2+ > Cd2+ > Zn2+, что вписывается в расширенный ряд сорбционного сродства элементов для катионитов с группами фосфоновых кислот [11].
Эти явления находят объяснения, которые сводятся к трем специфическим особенностям, присущим биполярным фосфорсодержащим ионитам с анионообменными группами в алкильном заместителе вместо кислотного гидроксила у атома фосфора. Первая особенность – увеличение степени кислотной силы и диссоциации фосфиновокислотной группы за счет электроноакцепторного эффекта катионного центра ≡N и ≡Р+, передаваемого через межцентровой фрагмент, причем степень передачи зависит от количества звеньев последнего (pKα1 фосфиновой группы практически не зависит от типа заместителей при атоме азота, но заметно возрастает с ростом числа метильных членов). Вторая особенность – образование и прочность связи катионного и фосфинового центра во внутрисолевой форме при формировании 3-, 4- и 5-членных циклов и возможность расщепления этой связи при конкуренции катиона поливалентного металла и катиона ионогенной группы; третья – повышение гидрофильности самих ионитов при введении амино- и фосфониевых групп в сочетании с гидроксильными.
Катионы металлов первого переходного ряда такими же однозвенными амфолитами с упорядоченным расположением кислотного и основного центра способны активно поглощаться только в слабокислых растворах (рН > 2), когда в фазе ионита создаются условия для максимальной ионизации кислотных групп и депротонизации аминогрупп. По всей вероятности, взаимодействие с ионогенными группами, изначально находящимися в цвиттер-ионном состоянии, происходит по реакции ионного обмена с замещением протона кислотного остатка и одновременно с образованием координационных связей с аминогруппой, сопровождающимся расщеплением бетаионовой структуры [12, 13]. Полностью исключать участие фосфорильного кислорода в комплексообразовании переходных металлов нет оснований, но, по нашему мнению, оно носит ограниченный характер, и если имеет место, то его значение второстепенно.
По сравнению с монофосфоновыми катионитами дифосфоновые сорбенты, содержащие дополнительные фосфоновые группы, соединенные с фосфиновыми подвижными α-, β-алкильными мостиками, обнаруживают очень высокие сорбционные характеристики по ионам индия и галлия (табл. 2). Такое влияние второй Р-группы, создающей с первой единый реакционный фрагмент, объяснимо с позиций комплексообразования, конформационных и стерических явлений, стабилизирующих комплексные формы [14, с. 256, 15]. Для ионов металлов, взаимодействующих с лигандными группами ионита, характерна координационная ненасыщенность. Вследствие особого строения сорбентов – нерастворимости и ограниченной подвижности макромолекулярного лигандоносителя – затруднено формирование комплексов со стехиометрией, обусловленной электронным строением иона металла. Катиониты типа СДФ с двумя близко расположенными фосфорильными группами, отделенными друг от друга алифатическими шарнирными звеньями, обеспечивают высокую подвижность гибкому реакционному фрагменту, легкость образования различных конфигураций, требуют существенно меньшего деформирования матрицы, способствуют формированию менее напряженных хелатных циклов и приводят к существованию более прочных мультидентатных, вероятно, квадрокоординационных комплексов (рис. 2).
Таблица 2.
Избирательность сорбции рассеянных элементов фосфиновокислотными амфолитами с основными заместителями и на основе макропористого сополимера стирола и ДВБ
| Марка ионита | Сорбируемость металлов, мг-экв/г | Коэффициенты распределения (Kр) | Коэффициенты избирательности (Kи) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In(III) | Ga(III) | Cu(II) | Zn(II) | Cd(II) | In(III) | Ga(III) | Cu(II) | Zn(II) | Cd(II) | In/Cu | In/Zn | In/Cd | Ga/Cu | Ga/Zn | Ga/Cd | |
| АФ-11 | 3.09 | 2.72 | 0.063 | 0.055 | 0.053 | 330.7 | 238.6 | 3.35 | 2.72 | 3.59 | 98.7 | 121.6 | 92.1 | 71.2 | 87.7 | 66.5 |
| АФ-13 | 2.76 | 2.34 | 0.054 | 0.080 | 0.050 | 250.8 | 176.0 | 2.84 | 1.96 | 3.35 | 88.3 | 128.0 | 74.9 | 62.0 | 89.8 | 52.5 |
| АФ-14 | 2.95 | 2.66 | 0.57 | 0.049 | 0.052 | 292.2 | 228.8 | 3.01 | 2.42 | 3.47 | 97.1 | 120.7 | 84.2 | 76.0 | 94.6 | 65.9 |
| АФ-21 | 2.89 | 2.07 | 0.05 | 0.046 | 0.044 | 273.3 | 142.3 | 2.67 | 2.27 | 2.99 | 102.4 | 120.4 | 91.4 | 53.3 | 62.7 | 47.6 |
| АФ-34 | 2.21 | 1.36 | 0.038 | 0.034 | 0.039 | 159.3 | 75.5 | 1.87 | 1.66 | 2.63 | 85.3 | 96.1 | 60.6 | 59.1 | 62.3 | 49.5 |
Рис. 2.
Зависимости сорбируемости ионов индия и галлия дифосфоновыми катионитами СДФ-12 (1, 2) и СДФ-20 (3, 4) и амфолитом АФ-11 (5, 6) от кислотности раствора: 1, 3 и 5 – индий (III); 2, 4 и 6 – галлий (III).

Образование в фазе ионита четырех- и трехкоординационных комплексов, более устойчивых благодаря дополнительному хелат-эффекту при формировании второго хелатного цикла:
требует определенной ориентации функциональных групп, что не всегда возможно и может приводить к значительным напряжениям в полимерной структуре сорбента. В этом случае энергетически более выгодно образование трехкоординированного комплекса. Уменьшение мобильности хелатогенных центров, например, в бензил- или фенилмонофосфоновых катионитах, приводит к предпочтительному образованию комплексов с меньшей координацией по макромолекулярным лигандам, что обязательно должно отразиться и отражается на величинах сорбируемости, особенно в сильнокислой области. В монокислотных ионитах формирование высококоординационных комплексов сопровождается участием в комплексообразовании групп различных полимерных цепей. Негативный момент при создании межцепных комплексов – увеличение “поперечных” связей в ионите и сжатие полимерной сетки, сопряженное со снижением проницаемости ионита, доступности функциональных групп и неполной реализацией потенциальных сорбционных возможностей.
Как правило, сорбционные показатели фосфоновых и фосфиновых ионитов с различными заместителями для изоэлектронных ионов индия (III) и галлия (III) сильно отличаются. В определенной мере исключение составляют дифосфоновые катиониты, для которых эти величины существенно сближаются, что предопределяет возможность их коллективного сорбционного извлечения. Значительное возрастание сорбируемости галлия катионитом с группировками α-1-фосфооксиэтилидендифосфоновой кислоты (СДФ-12) согласуется с тем, что мономерная форма хелатная и характеризуется одной из самых высоких констант устойчивости растворимых в водных растворах ацидокомплексов по сравнению с другими фосфорсодержащими соединениями [16, гл. 5.2], к тому же выгодно отличается по прочности образующихся комплексов от ионов меди (II) [17] и цинка (II) [18]. И это в определенной мере свидетельствует о сохранении специфичности комплексообразования оксиэтилидендифосфонового лиганда, закрепленного на полимерном носителе.
При аналогии строения электронных контуров индия и галлия главными видимыми признаками, позволяющими понять причины большей склонности катионов индия к реализации своих притязаний на ионно-координационное взаимодействие с фосфорсодержащими группами сорбента по сравнению с галлием остаются благоприятные размеры атома индия (0.92 Å [19, с. 66] против 0.62 Å [16, с. 45]) и его предрасположенность к электронной поляризации.
Другие структурно-подобные катиониты с дифосфоновыми группами (известные под маркой Diphonix) также проявляют повышенные сорбционные показатели по ионам индия (III) и галлия (III) в кислых растворах [20].
Таким образом, введение к атому фосфора фосфинитного центра второй фосфоновой группы, увеличивающей общую дентатность функционального фрагмента сорбента, оказывает существенное положительное влияние на сорбционную емкость по рассеянным элементам, особенно по ионам галлия (III). А общее преимущество подобных фосфиновокислотных ионитов – возможность широкого варьирования заместителей при атоме фосфора, что позволяет изменять силу координационного центра и пространственные факторы вблизи него, оказывающие большое влияние на комплексообразующую способность лигандов, закрепленных на полимерной основе. В условиях идентичности пористости матрицы решающее влияние на сорбционные характеристики фосфиновокислотных катионитов по ионам индия и галлия принадлежит координационным и гидратационным свойствам заместителей в ионогенных группах.
Список литературы
Радионов Б.К., Лейкин Ю.А., Смирнов А.Л., Свирский И.М. // Журн. физ. химии. 2019. Т. 93. № 12.
Дедов В.Б., Калиниченко Б.С., Лейкин Ю.А. и др. К вопросу о возможности хроматографического разделения кюрия и калифорния на фосфор- и фосфоразотсодержащих ионитах / Препринт института атомной энергии им И.В. Курчатова. ИАЭ-3303/13. М., 1980. 37 с.
Лейкин Ю.А. Физико-химические основы синтеза полимерных сорбентов. М.: БИНОМ. Лаборатория знаний. 2011. С. 413.
Носкова М.П., Радионов Б.К., Молочников Л.С. и др. // Журн. физ. химии. 1984. Т. 58. № 5. С. 1235.
Maeda H., Egawa H. // J. Appl, Polymer Sci. V. 42. № 3. P. 737.
Шварц А.Л., Молочников Л.С., Иванова Т.М. и др. // Журн. физ. химии. 1983. Т. 57. № 9. С. 2292.
Молочников Л.С., Радионов Б.К., Носкова М.П. и др. // Там же. 1990. Т. 64. № 4. С. 987.
Лейкин Ю.А., Амелина Ж.С., Коршак В.В. // Высокомолекулярные соединения. 1976. Т. 18 А. № 2. С. 364.
Балакин В.М., Тэслер А.Г., Балакин С.М. и др. // Там же. 1976. Т. 18 Б. № 6. С. 423.
Балакин В.М., Драницина И.В., Холманиский Ю.Б. и др. // Журн. прикл. химии. 1981. Т. 54. № 4. С. 781.
Marhol M., Beranová H., Cheng K.L. // J. Radioanalyt. Chem. 1974. V. 21. № 1. P. 177.
Колосова И.Ф., Тарасова Т.И., Лейкин Ю.А. и др. // Коорд. химия.1982. Т. 8. № 9. С. 1193.
Колосова И.Ф., Тарасова Т.И., Лейкин Ю.А. и др. // Там же. 1982. Т. 8. № 11. С. 1502−1509.
Салдадзе К.М., Копылова-Валова В.Д. Комплексообразующие иониты (комплекситы). М.: Химия, 1980. 332 с.
Салдадзе К.М., Копылова-Валова В.Д. // Журн. аналит. химии. 1972. Т. 27. № 5. С. 956.
Радионов Б.К., Мальцев Г.И. Галлий в водных растворах. Химия редких и рассеянных элементов // Germany, Saarbrücker: LAP LAMBERT Academic Publishig. 2014. 313 с.
Вассерштейн Ш.Е., Нгуен Ван Нам // Журн. неорг. химии. 1973. Т. 18. № 4. С. 1028.
Васильев В.П., Козловский Е.В., Марьина Т.Б., Летягина Г.И. // Там же. 1987. Т. 32. № 8. С. 1916.
Радионов Б.К., Мальцев Г.И. Индий в водных растворах. Химия редких и рассеянных элементов // Germany, Saarbrücker: LAP LAMBERT Academic Publishig. 2014. 352 с.
Trochimczuk A.W., Horwitz E.P., Alexandratos S.D. // Separat. Sci. Technol. 1994. V. 29. № 4. P. 543.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии