

Журнал физической химии, 2020, T. 94, № 4, стр. 617-623
Модель электрофоретического поведения комплексов металлов
Е. Л. Лебедева a, b, *, Л. К. Неудачина a
a Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
Екатеринбург, Россия
b Уральский научно-исследовательский институт метрологии
Екатеринбург, Россия
* E-mail: ElenaLebedeva@urfu.ru
Поступила в редакцию 10.05.2019
После доработки 11.06.2019
Принята к публикации 18.06.2019
Аннотация
Предложена математическая модель, описывающая поведение комплексных соединений металлов в процессе их разделения методом капиллярного зонного электрофореза. Разработанная модель позволяет объяснять возможность качественного и количественного определения ионов металлов в виде их комплексов с органическими реагентами; предсказывать порядок миграции комплексов и оценивать времена их миграции; выбирать наиболее благоприятные условия для их определения. Показано, что для лабильных комплексов возможность наблюдения их пика определяется величиной условной константы устойчивости. Для определения малоустойчивых заряженных комплексов наиболее благоприятны соответствующая полярность и высокая скорость электроосмотического потока, при этом величины напряжения и ввода пробы следует выбирать экспериментально, учитывая как селективность разделения, так и чувствительность определения. Рассчитанные величины времен миграции и вид градуировочных графиков для комплексов различных металлов с органическими хелатообразующими реагентами хорошо согласуются с результатами экспериментов.
При определении ионов металлов в растворах методом капиллярного зонного электрофореза (КЗЭ) с УФ-детектированием их обычно переводят в форму комплексных соединений для повышения чувствительности и селективности разделения. Чаще всего для этой цели используют органические хелатообразующие реагенты, которые добавляют в раствор пробы перед анализом или в фоновый электролит. Эффективная электрофоретическая подвижность ионов металлов зависит от равновесий между аквакомплексами ионов металлов М, ионами или молекулами реагента L, комплексными частицами, компонентами фонового электролита и др. Таким образом, рассчитать величины подвижностей ионов металлов, не прибегая к эксперименту, бывает очень сложно. Поэтому выбор комплексообразующего реагента и условий электрофоретического анализа, обеспечивающих оптимальное разделение пиков комплексов, осуществляют в большинстве случаев последовательным экспериментальным варьированием соответствующих параметров.
Предложены приближенные эмпирические модели, связывающие величину электрофоретической подвижности комплекса с его составом и характеристиками иона металла и лиганда, пользуясь которыми, можно предсказать времена миграции других комплексов сходного строения [1, 2]. Однако данный подход применим лишь для небольшого количества однотипных комплексов и зачастую затрудняется недоступностью или недостаточной точностью исходных данных. Обычно рассматривают только один параметр, влияющий на подвижность (μi) комплекса MLi – термодинамическую константу его устойчивости [3–6]. Эффективная подвижность иона металла в этом случае является усредненной подвижностью всех n металлсодержащих комплексов [7–13]:
Если комплексообразующий реагент является слабой кислотой, то, зная константы равновесий протонирования, можно оценить оптимальную концентрацию его в фоновом электролите и значение рН, обеспечивающие наибольшую разность эффективных подвижностей комплексов разделяемых металлов [9, 14]. Если возможно образование моноядерных комплексов различного состава, в качестве независимой переменной для определения величины подвижности комплекса используют среднее число лигандов, связанных с одним ионом металла [15–17].
Перечисленные выше методы применяются, когда комплексообразующий реагент входит в состав фонового электролита. Известно несколько работ, посвященных исследованию факторов, влияющих на подвижность комплексов при предварительном комплексообразовании [18–23]. В этом случае не все комплексы, содержащиеся в пробе, можно фиксировать в виде пиков на электрофореграмме (ЭФГ). Этот факт объясняют невысокой термодинамической устойчивостью комплекса в сочетании с его кинетической лабильностью, что приводит к диссоциации комплекса в процессе анализа [18, 21, 23]. Критерий, согласно которому, данный комплекс можно считать кинетически лабильным или инертным, подробно обсуждается в [22]. В работах [18–22] уточняется, что причиной диссоциации является разная скорость миграции комплекса и лиганда, что приводит к их физическому разделению в капилляре. Однако в литературе нами не было найдено критериев, позволяющих оценить возможность количественного определения комплекса методом КЗЭ или выбрать оптимальные условия разделения ионов, не прибегая к многочисленным и трудоемким экспериментам.
Цель данной работы – установить количественные критерии, позволяющие объяснять и предсказывать возможность определения комплексов ионов металлов с органическими реагентами методом КЗЭ, а также использовать полученные данные для разработки методик совместного определения ионов металлов в реальных объектах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали реактивы квалификации не ниже “х. ч.”. Исходные растворы готовили растворением навесок соответствующих веществ в воде. Рабочие растворы готовили последовательным разбавлением исходных.
Новый капилляр готовили к работе последовательными 30-минутными промывками 1 моль/дм3 раствором HCl, деионизованной водой, 0.5 моль/дм3 раствором NaOH, затем водой и фоновым электролитом. Между измерениями капилляр промывали фоновым электролитом в течение 3 мин. В качестве фонового электролита использовали фосфатный или боратный буферный растворы, которые готовили в день использования.
Регистрацию ЭФГ и спектров поглощения растворов проводили на системе капиллярного электрофореза “Капель 105М” (ООО “Люмэкс”, Россия) c немодифицированным кварцевым капилляром (внутренний диаметр 75 мкм, эффективная длина 50 см, общая длина 60 см) и УФ-детектором. Измерение концентрации ионов металлов в растворах осуществляли на атомно-абсорбционном спектрометре Solaar M6 (ThermoElectron, США) с пламенной атомизацией, а также на атомно-эмиссионном спектрометре c индуктивно связанной плазмой iCAP-6500 Duo (Thermo Scientific, США). Значение рН растворов контролировали с помощью лабораторного анализатора жидкости “АНИОН 4100” (ООО НПП “Инфраспак-Аналит”, Россия) со стеклянным комбинированным электродом ЭСК-10601/7.
Для приготовления всех растворов использовали деионизованную воду удельным сопротивлением не менее 18.2 МОм см (система высокой очистки воды Milli-Q А10 Academic (Millipore, США)).
Идентификацию пиков на ЭФГ проводили методом добавок или по временам миграции. Для расчета равновесных концентраций частиц в растворах использовали программы “Hyperquad Simulation and Speciation” (Protonic Software), “ChemEQL v. 3.1”, а также систему компьютерной алгебры “Maple 16.01”. В качестве источников констант равновесия использовали литературные данные и программы “Mini-SCDatabase” и “NIST Critically Selected Stability Constants of Metal Complexes”.
МЕТОДИКА РАСЧЕТОВ
Константа устойчивости комплекса ML связана с равновесными концентрациями ионов металла, лиганда и комплекса выражением
Данные частицы могут участвовать в посторонних взаимодействиях с компонентами пробы и фонового электролита, и тогда для характеристики устойчивости комплекса можно использовать величину условной константы устойчивости
где $C_{{{\text{ML}}}}^{'}$ – суммарная концентрация частиц, в которых металл связан c реагентом; $C_{{\text{M}}}^{'}$ – суммарная концентрация частиц, в которых металл не связан с реагентом; а $C_{{\text{L}}}^{'}$ – суммарная концентрация частиц, в которых реагент не связан с металлом. Величину Kусл можно рассчитать, зная общие концентрации металла, лиганда и компонентов фонового электролита, его рН и константы равновесий, возможных в системе. Проведенные нами расчеты показывают, что в условиях КЗЭ (постоянный состав и кислотность фонового электролита, CM$ \ll $ CL ≤ 0.01 моль/дм3) величина Kусл может считаться постоянной в широком интервале концентраций М и L.Пусть в капилляр введена зона пробы длиной lВП, содержащей ионы металла М общей концентрацией CM0 и комплексообразующий реагент L общей концентрацией CL0. Равновесные концентрации до начала анализа [M]0, [L]0 и [ML]0 можно рассчитать по уравнениям:
(1)
${{K}_{{{\text{уст}}}}} = \frac{{{{{[{\text{ML}}]}}_{0}}}}{{({{C}_{{{\text{M}}0}}} - {{{[{\text{ML}}]}}_{0}})({{C}_{{{\text{L0}}}}} - {{{[{\text{ML}}]}}_{0}})}},$При подаче высокого напряжения ионы М и L начинают двигаться относительно зоны пробы в противоположных направлениях со скоростями ${{{v}}_{{\text{M}}}}$ и ${{{v}}_{{\text{L}}}}$ соответственно (рис. 1), что приводит к диссоциации комплекса. Приняв некоторые допущения:
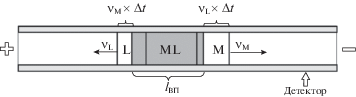
1) скорость движения иона в процессе анализа остается постоянной;
2) концентрации компонентов в границах зоны достаточно быстро выравниваются за счет диффузии; диффузионным размыванием зон при этом можно пренебречь;
3) комплекс достаточно лабилен, так что в любой момент в границах зоны существует химическое равновесие между частицами М, L и ML, можно рассчитать величину х, на которую уменьшится равновесная концентрация комплекса в зоне за малый промежуток времени Δt, решив относительно х уравнение:
(4)
$\begin{gathered} {{K}_{{{\text{усл}}}}} = \\ = \frac{{[{\text{ML}}] - x}}{{\left( {[{\text{M}}] - \frac{{{{{v}}_{{\text{M}}}}\Delta t}}{{{{l}_{{{\text{вп}}}}}}}[{\text{M}}] + x} \right)\left( {[L] - \frac{{{{{v}}_{{\text{L}}}}\Delta t}}{{{{l}_{{{\text{вп}}}}}}}[L] + x} \right)}}, \\ \end{gathered} $Таким образом, величина Kусл может быть принята в качестве основного критерия возможности электрофоретического определения комплексов ионов металлов различного состава [24].
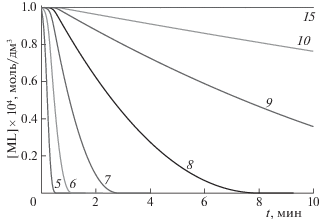
Расчеты, проведенные для модельных систем (рис. 2), показывают, что даже при довольно высокой концентрации иона металла в пробе количественное его определение может представлять трудности, если комплекс недостаточно устойчив в выбранных условиях анализа. Так, комплексы с lg Kусл < 7, скорее всего, не образуют отдельных пиков, при этом увеличение содержания металла в пробе не приводит к уменьшению площади пика реагента. Комплексы с lg Kусл = 7–11 могут проявляться на ЭФГ в виде отдельных размытых пиков; в этом случае требуется тщательный выбор оптимальных условий анализа. Кроме того, расчеты показывают, что для малоустойчивых комплексов градуировочные графики могут иметь не прямолинейную, а вогнутую форму [24].
Рис. 2.
Зависимости равновесной концентрации комплекса ML от времени при различной величине логарифма условной константы устойчивости комплекса; CM0 = 1 × 10–4 моль/дм3, CL0 = 1 × 10–4 моль/дм3, lВП = 2.5 мм, ${{{v}}_{{\text{M}}}}$ = 0.25 см/с, ${{{v}}_{{\text{L}}}}$ = 0.05 см/с.
Изучение влияния различных параметров на величину [ML] показывает, что для чувствительного определения малоустойчивых комплексов наиболее благоприятными условиями будут следующие:
1) полярность, соответствующая заряду комплекса, и возможно более высокая скорость электроосмотического потока (ЭОП);
2) состав фонового электролита (природа буферного раствора, концентрация и рН) обеспечивающий максимальные величины Kусл, минимальные ${{{v}}_{{\text{M}}}}$ и ${{{v}}_{{\text{L}}}}$ и при этом приемлемую скорость анализа;
3) СM < CL, при этом соотношение концентраций не имеет большого значения;
4) объем введенной пробы достаточно велик, чтобы обеспечить максимальную чувствительность, но при этом избежать перегрузки капилляра и достичь селективности разделения зон разных комплексов.
Часто можно принять, что подвижности аквакомплексов многих металлов близки, поэтому для описания поведения целого ряда комплексов на практике достаточно измерить подвижность реагента, наиболее устойчивого комплекса и иона соответствующего металла.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Проведенные нами исследования электрофоретического поведения комплексов металлов с некоторыми органическими хелатообразующими реагентами подтвердили, что пики комплексов не всегда появляются на ЭФГ даже при большой их концентрации в пробе. Так, например, нам не удалось наблюдать пики, соответствующие комплексам ионов меди, кобальта и никеля с реагентами класса N-арил-3-аминопропионовых кислот (ААПК, рис. 3). Данные соединения обладают селективностью к ионам меди(II) [25–27] и применяются для их избирательного спектрофотометрического [28, 29], потенциометрического [28] и люминесцентного [30] определения в различных объектах. Расчет Кусл для наиболее прочных комплексов с Cu(II) (рис. 4) показывает, что все они недостаточно устойчивы в условиях анализа, что затрудняет их определение методом КЗЭ.

Рис. 4.
Зависимости логарифма условной константы устойчивости комплексов реагентов RI –RV с Cu(II) от рН раствора.

Отсутствие пиков наблюдалось и для некоторых комплексов с ЭДТА. Сравнение условных констант устойчивости этилендиаминтетраацетатных комплексов (табл. 1) в боратном фоновом электролите при рН 9.2 позволяет объяснить, почему на ЭФГ не удалось наблюдать пики комплексов Al, Hg(II) и Ag(I): их устойчивость недостаточно высока в условиях анализа.
Таблица 1.
Условные константы устойчивости (lg Kусл) комплексов М-ЭДТА
| Металл | Ag(I) | Al | Bi | Cd | Co(II) | Cu(II) | Fe(III) | Hg(II) | Mn(II) | Ni(II) | Pb(II) | Zn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| рН 9.2 | 6.27 | 6.72 | 18.05 | 14.84 | 15.19 | 14.05 | 10.16 | 8.83 | 14.55 | 17.31 | 21.96 | 15.89 |
| pH 4.5 | 1.79 | –0.59 | 20.60 | 5.02 | 9.03 | 10.35 | 12.41 | 11.55 | 8.06 | 11.07 | 11.90 | 9.21 |
В то же время нами экспериментально показано, что комплексы, обладающие наибольшей устойчивостью в фосфатном буферном растворе при рН 4.5 (Bi(III), Fe(III), Pb(II) и Cu(II)), возможно определять количественно с достаточно высокой чувствительностью при следующих условиях: 0.1 моль/дм3 фосфатный буферный раствор с 1 × 10–4 моль/дм3 гидроксида цетилтриметиламмония в качестве фонового электролита, напряжение –20 кВ, температура 25°С, длина волны детектирования 260 нм, гидродинамический ввод пробы 250 мбар c. Однако селективность разделения комплексов М-ЭДТА в большинстве случаев невелика, поэтому для одновременного определения четырех указанных металлов мы использовали диглицилглицин в качестве комплекс-селектора [31].
Использование предлагаемой модели позволило выбрать условия для селективного определения ионов Ni(II) и Co(II), комплексы которых с ЭДТА не разделяются ни в кислой, ни в щелочной среде. Для этого в качестве комплексообразующего реагента был выбран 1,10-фенантролин (Phen). Данный реагент использовали для определения ионов Fe(II), Cu(II), Zn(II), Co(II), Ni(II), Mn(II) и Cd(II) методом КЗЭ в различных объектах [32–39], однако примеров одновременного определения Co(II) и Ni(II) и систематического изучения электрофоретического поведения комплексов других тяжелых металлов нами не было найдено.
Чтобы исследовать мешающее влияние тяжелых металлов, которые могут присутствовать в пробе, также изучали поведение комплексов Cu(II), Fe(II), Fe(III), Mn(II), Zn(II), Cd(II) и Pb(II). Исходя из рассчитанных значений Kусл для разделения был выбран фоновый электролит на основе фосфатного буферного раствора с рН 8.0. Поскольку все комплексы положительно заряжены, разделение проводили при положительной полярности. Длина волны детектирования была выбрана равной 190 нм, поскольку при этой длине волны наблюдается наибольшее светопоглощение комплексов. Величины напряжения и ввода пробы выбирали экспериментально так, чтобы обеспечить наилучшую чувствительность определения, достаточное разрешение пиков и приемлемую скорость анализа.
Экспериментальные исследования показали, что электрофоретическое поведение комплексов соответствует ожидаемому (рис. 5): чем выше значение Kусл комплекса, тем больше вероятность появления пика на ЭФГ и его количественного определения. Для комплексов Ni(II) и Co(II) наблюдаются четкие, хорошо воспроизводимые пики, площади которых прямо пропорциональны концентрации металла в довольно широком интервале (табл. 2). Комплексы Zn(II) и Cd(II) появляются на ЭФГ в виде ступенчатых размытых пиков, которые трудно разметить; пределы обнаружения этих металлов довольно высоки, а диапазон линейности градуировочного графика узок. Комплекс Cu(II) характеризуется чрезвычайно размытым, практически непригодным для количественного анализа пиком. Присутствие ионов Mn(II) в пробе приводит только к изменению формы пика реагента; пики, соответствующие комплексам Fe(III) и Pb(II), на ЭФГ не появляются [24].
Рис. 5.
ЭФГ растворов, содержащих 5 × 10–4 моль/дм3 Phen (1) и 1.6 × 10–4 моль/дм3 ионов Pb(II) (2), Cd(II) (3), Cu(II) (4), Zn(II) (5), Fe(III) (6), Mn(II) (7), Fe(II) (8), Ni(II) (9), Co(II) (10). Фоновый электролит: 0.025 моль/дм3 фосфатный буферный раствор, рН 8.0. U = +15 кВ, λ = 190 нм, t = 20°C, ввод пробы 300 мбар с (для наглядности ЭФГ смещены по оси времени).

Таблица 2.
Определение комплексов некоторых тяжелых металлов с Phen (все условия как на рис. 5)
| Металл | lg Kусл | μ × 108, м2/(В с) | с1, моль/дм3 | с2 × 105, моль/дм3 | с3 × 105, моль/дм3 |
|---|---|---|---|---|---|
| Ni(II) | 14.80 | 2.80 ± 0.10 | 1.0 × 10–6–5.0 × 10–4 | 1.7 | 5.9 |
| Co(II) | 10.52 | 2.53 ± 0.16 | 5.0 × 10–6–5.0 × 10–4 | 1.4 | 2.4 |
| Zn(II) | 7.95 | 1.13 ± 0.25 | 4.0 × 10–5–1.6 × 10–4 | 2.9 | 7.8 |
| Cu(II) | 8.50 | 0.92 ± 0.09 | 8.0 × 10–5–2.5 × 10–4 | 10 | 22 |
| Fe(II) | 6.41 | 2.40 ± 0.03 | 4.0 × 10–5–1.6 × 10–4 | 3.2 | 6.8 |
| Cd(II) | 6.38 | 0.18 ± 0.08 | 4.0 × 10–5–1.6 × 10–4 | – | – |
| Fe(III) | 4.94 | – | – | – | – |
| Mn(II) | 3.74 | – | – | – | – |
| Pb(II) | 3.43 | – | – | – | – |
Примечания: μ – подвижность (число параллельных определений 3, доверительная вероятность 0.95), с1 – диапазон линейности, с2 – предел обнаружения (рассчитан как концентрация, соответствующая отношению сигнал/шум = 3), с3 – предел определения (рассчитан как концентрация, соответствующая отношению сигнал/шум = 10).
Кроме того, модель позволяет оценить время появления комплекса на ЭФГ, поскольку его эффективная подвижность пропорциональна средней доле металла, связанного в комплекс, за все время анализа. Сравнение рассчитанного времени миграции комплекса с экспериментальным показывает достаточно хорошее совпадение (табл. 3).
Таблица 3.
Экспериментальные и рассчитанные времена миграции комплексов М-Phen
| Металл | ${{t}_{{\text{L}}}}$, мин | ${{t}_{{{\text{ML, эксп}}}}}$, мин | ${{t}_{{{\text{ML, расч}}}}}$, мин |
|---|---|---|---|
| Ni(II) | 6.55 ± 0.22 | 4.86 ± 0.14 | 4.91 ± 0.14 |
| Co(II) | 6.63 ± 0.05 | 4.90 ± 0.06 | 4.94 ± 0.04 |
| Zn(II) | 6.71 ± 0.11 | 5.26 ± 0.18 | 5.01 ± 0.08 |
| Cd(II) | 10.80 ± 0.13 | 10.18 ± 0.18 | 10.37 ± 0.13 |
| Mn(II) | 8.68 ± 0.18 | 8.68 ± 0.18 | 8.68 ± 0.18 |
| Fe(II) | 8.81 ± 0.21 | 5.36 ± 0.23 | 5.02 ± 0.12 |
Исследование поддержано программой 211 Правительства Российской Федерации, соглашение № 02.A03.21.0006.
Список литературы
Timerbaev A.R., Semenova O.P., Petrukhin O.M. // J. Chromatogr. A. 2002. V. 943. № 2. P. 263.https://doi.org/10.1016/S0021-9673(01)01462-5
Motomizu S., Oshima M., Matsuda S. et al. // Anal. Sci. 1992. V. 8. № 5. P. 619.https://doi.org/10.2116/analsci.8.619
Timerbaev A.R., Buchberger W., Semenova O.P. et al. // J. Chromatogr. A. 1993. V. 630. № 1–2. P. 379.https://doi.org/10.1016/0021-9673(93)80475-N
Chen Z., Naidu R. // J. Chromatogr. A. 2002. V. 966. № 1–2. P. 245.https://doi.org/10.1016/S0021-9673(02)00741-0
Urbanek M., Delaunay N., Michel R. et al. // Electrophoresis. 2007. V. 28. № 20. P. 3767.https://doi.org/10.1002/elps.200700180
Clark S.B., Friese J.I. // J. Radioanal. Nucl. Chem. 2009. V. 282. № 2. P. 329.https://doi.org/10.1007/s10967-009-0324-z
Swaile D.F., Sepaniak M.J. // Anal. Chem. 1991. V. 63. № 2. P. 179.https://doi.org/10.1021/ac00002a017
Motomizu S., Mori N., Kuwabara M. et al. // Anal. Sci. 1994. V. 10. № 1. P. 101.https://doi.org/10.2116/analsci.10.101
Wang T., Li S.F.Y. // J. Chromatogr. A. 1995. V. 707. № 2. P. 343.https://doi.org/10.1016/0021-9673(95)00321-D
Ito K., Hirokawa T. // J. Chromatogr. A. 1996. V. 742. № 1–2. P. 281.https://doi.org/10.1016/0021-9673(96)00224-5
Padarauskas A., Naujalis E., Norkus E. et al. // Chromatographia. 2000. V. 52. № 7–8. P. 509.https://doi.org/10.1007/BF02535728
Foret F. // Electrophoresis. 2009. V. 30. № S1. P. S34.https://doi.org/10.1002/elps.200900004
Sursyakova V.V., Rubaylo A.I. // Electrophoresis. 2018. V. 39. № 8. P. 1079.https://doi.org/10.1002/elps.201700378
Yang Q., Smeyers-Verbeke J., Wu W. et al. // J. Chromatogr. A. 1994. V. 688. № 1–2. P. 339.https://doi.org/10.1016/0021-9673(94)00800-0
Erim F.B., Boelens H.F.M., Kraak J.C. // Anal. Chim. Acta. 1994. V. 294. № 2. P. 155.https://doi.org/10.1016/0003-2670(94)80190-8
Shi Y., Fritz J.S. // J. Chromatogr. A. 1993. V. 640. № 1–2. P. 473.https://doi.org/10.1016/0021-9673(93)80217-V
Conradi S., Vogt C., Wittrisch H. et al. // J. Chromatogr. A. 1996. V. 745. № 1–2. P. 103.https://doi.org/10.1016/0021-9673(96)00268-3
Bürgisser C.S., Stone A.T. // Environ. Sci. Technol. 1997. V. 31. № 9. P. 2656.https://doi.org/10.1021/es970080f
Macka M., Nesterenko P., Andersson P. et al. // J. Chromatogr. A. 1998. V. 803. № 1–2. P. 279.https://doi.org/10.1016/S0021-9673(97)01226-0
Liu B.-F., Liu L.-B., Chen H. et al. // Fresenius J. Anal. Chem. 2001. V. 369. № 2. P. 195.https://doi.org/10.1007/s002160000604
Himeno S., Kitano E., Morishita K. // Anal. Sci. 2007. V. 23. № 8. P. 959.https://doi.org/10.2116/analsci.23.959
Carbonaro R.F., Stone A.T. // Anal. Chem. 2005. V. 77. № 1. P. 155.https://doi.org/10.1021/ac048860b
Fung Y., Lau K. // Electrophoresis. 2001. V. 22. № 11. P. 2192.https://doi.org/10.1002/1522-2683(20017)22:11<2192::AID-ELPS2192>3.0.CO;2-A
Лебедева Е.Л. Электрофоретическое разделение и определение ионов металлов в виде комплексов с органическими реагентами: Дис. … канд. хим. наук. Екатеринбург: Урал. федер. ун-т, 2014. 167 с.
Скорик Ю.А., Неудачина Л.К., Вшивков А.А. и др. // Журн. неорган. химии. 2001. Т. 46. № 1. С. 143.
Скорик Ю.А., Неудачина Л.К., Вшивков А.А. // Журн. общ. химии. 1999. Т. 69. № 2. С. 296.
Скорик Ю.А., Подберезская Н.В., Романенко Г.В. и др. // Журн. неорган. химии. 2003. Т. 48. № 2. С. 250.
Неудачина Л.К., Осинцева Е.В., Скорик Ю.А. и др. // Журн. аналит. химии. 2005. Т. 60. № 3. С. 271.
Скорик Ю.А., Неудачина Л.К., Коротовских Е.В. и др. // Зав. лаб. 2001. Т. 67. № 5. С. 15.
Осинцева Е.В., Неудачина Л.К., Амфитеатрова А.В. и др. // Зав. лаб. 2006. Т. 72. № 9. С. 11.
Неудачина Л.К., Лебедева Е.Л. // Аналитика и контроль. 2014. Т. 18. № 4. С. 458.https://doi.org/10.15826/analitika.2014.18.4.013
Xu J., Ma Y. // J. Microcol. Sep. 1996. V. 8. № 2. P. 137.https://doi.org/10.1002/(SICI)1520-667X(1996)8:2< 137::AID-MCS7>3.0.CO;2-2
Xu J., Che P., Ma Y. // J. Chromatogr. A. 1996. V. 749. № 1−2. P. 287.https://doi.org/10.1016/0021-9673(96)00457-8
Schäffer S., Gareil P., Dezael C., Richard D. // J. Chromatogr. A. 1996. V. 740. № 1. P. 151.https://doi.org/10.1016/0021-9673(96)00121-5
Pozdniakova S., Padarauskas A. // Analyst. 1998. V. 123. № 7. P. 1497.https://doi.org/10.1039/a800892b
Pozdniakova S., Padarauskas A., Schwedt G. // Anal. Chim. Acta. 1997. V. 351, № 1−3. P. 41.https://doi.org/10.1016/S0003-2670(97)00331-0
Dabek-Zlotorzynska E., Aranda-Rodriguez R., Buykx S. // Anal. Bioanal. Chem. 2002. V. 372. № 3. P. 467.https://doi.org/10.1007/s00216-001-1114-9
Yokoyama T. // Anal. Chim. Acta. 1998. V. 364. № 1−3. P. 75.https://doi.org/10.1016/S0003-2670(98)00153-6
Naujalis E., Padarauskas A. // J. Chromatogr. A. 2002. V. 977. № 1. P. 135.https://doi.org/10.1016/S0021-9673(02)01350-X
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии