

Журнал физической химии, 2020, T. 94, № 5, стр. 747-753
Квантово-химическое моделирование реакций согласованного распада циклов
Н. С. Емельянова a, b, *, Т. С. Покидова a
a Российская академия наук, Институт проблем химической физики
142432 Московской области, Черноголовка, Россия
b Московский государственный университет имени М.В. Ломоносова, Факультет фундаментальной физико-химической инженерии
119991 Москва, Россия
* E-mail: n_emel@mail.ru
Поступила в редакцию 17.07.2019
После доработки 17.07.2019
Принята к публикации 17.09.2019
Аннотация
Проведено квантово-химическое моделирование ретро-реакций Дильса–Альдера на примере циклов различного строения – с алкильными заместителями и с гетероатомами в цикле. С помощью топологического анализа функции распределения электронной плотности (QTAIM) проведен топологический анализ переходных состояний и промежуточных соединений вблизи них. На основании полученных данных о строении переходных состояний и механизме реакции создан алгоритм расчета энергии активации согласованного распада цикла в рамках метода пересекающихся парабол (М3ПП). Показано влияние строения исходного цикла на протекание реакции.
Газофазные реакции молекулярного распада органических соединений составляют важную область теоретической и прикладной химии [1–4]. К ним можно отнести и реакции распада шестичленных циклов в газовой фазе при высокой температуре (>600 K) [5–13]:
Здесь X = C, N, O, S; Y = C или O. Реакции протекают путем согласованной перегруппировки и распада связей через шестичленное переходное состояние (ПС) с образованием сопряженных диенов и соответствующего диенофила [5–8], являясь, таким образом, реакциями, обратными диеновому синтезу Дильса–Альдера.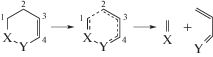
Для анализа реакций согласованного распада различных классов соединений с циклическим ПС [14–19] весьма успешно использовались такие методы исследования как модели пересекающихся парабол (МПП) и (М3ПП) [20–22], позволяющие построить полуэмпирический алгоритм расчета и рассчитать энергии активации (Е) и константы скорости (k) реакций, выявить и оценить факторы, влияющие на величину Е. В их рамках ПС можно рассматривать как точку пересечения двух невозмущенных потенциальных кривых – парабол, из которых одна характеризует потенциальную энергию валентного колебания рвущейся связи, другая – образующейся. Для более корректного выбора такой пары необходимо как можно точнее знать геометрию и электронное строение переходного состояния. С этой задачей хорошо справляется квантово-химическое моделирование [23]. Топологический анализ функции распределения электронной плотности (QTAIM) [24] дает нам еще более точные данные об электронном строении ПС, возможность определить тип связей и их энергию в нем. Для исследуемых реакций важно знать также их путь, поэтому цель данной работы – изучение топологических характеристик связей вдоль координаты реакции, факторов, влияющих на них и сопоставление результатов расчета энергии активации с помощью модели пересекающихся парабол и квантово-химического моделирования с использованием метода функционала плотности (DFT).
МЕТОДЫ АНАЛИЗА И РАСЧЕТА
Квантово-химические расчеты проводились по программе Gaussian 09 версия D [25] с помощью гибридного функционала B3LYP и базисного набора 6-311++G**. Расчет частот в оптимизированных геометриях исходных и конечных продуктов показал отсутствие мнимых частот, поэтому все оптимизированные геометрии соответствуют минимумам энергии. Для всех полученных переходных состояний был проведен расчет частот, который показал наличие мнимой частоты. Для установления истинности переходных состояний были проведены IRC-расчеты, которые использовались и для анализа профилей поверхности потенциальной энергии вблизи переходного состояния.
Для анализа волновой функции методами теории AIM использовался программный пакет AIMAll (Version 15.05.18) [26]. Расчеты волновых функций и топологических характеристик распределения электронной плотности выполнены в тех же приближениях, что и оптимизация переходных состояний. Энергии связей рассчитывались по известной корреляционной формуле [27]:
где EA–B – энергия связи, ${{{v}}_{{\text{e}}}}(r)$ – плотности потенциальной энергии в критической точке этой связи.Полуэмпирический метод пересекающихся парабол М3ПП проводился по методике, описанной в [21].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Квантово-химические расчеты
Теоретическое исследование проводилось на примере восьми ретро-реакций Дильса–Альдера. Для каждой из рассматриваемых реакций было найдено ПС, которое было проанализировано с помощью метода QTAIM (рис. 1). Это – распад шестичленных непредельных циклов с одной двойной связью (ПС1–ПС4), циклов, которые могут содержать гетероатом в β-положении от двойной связи (ПС5, ПС7 и ПС8) и непосредственно рядом с двойной связью (ПС6) (см. рис. 1).
Рис. 1.
Молекулярные графы оптимизированных геометрий ПС, полученные из расчетов в рамках теории QTAIM [19].
Как видно из рис. 1, все ПС представляют собой неплоские шестичленные циклы. В случае распада циклогексена это – симметричное ПС, при введении заместителей или гетероатомов, оно становится несимметричным. По своей геометрии ПС ближе к конечным продуктам реакции, т.е. ПС являются “поздними” и нам следует ожидать значительного энергетического барьера реакции. Несмотря на значительное расстояние между атомами разрывающихся связей С–С (ПС1–ПС4) или С–X, где X = O, S, N (ПС5–ПС8), между ними имеются точки типа (3, –1), в которых градиент волновой функции равен нулю, так называемые критические точки связи (КТ). Их наличие свидетельствует о том, что между этими атомами имеется химическая связь. Анализ критических точек даст нам возможность судить о ее прочности и типе (табл. 1).
Таблица 1.
Параметры разрывающихся связей в переходных состояниях циклов из метода QTAIM
| Параметр | Связь | ПС1 | ПС2 | ПС3 | ПС4 | ПС5 | ПС6 | ПС7 | ПС8 |
|---|---|---|---|---|---|---|---|---|---|
| ρ(r) | С–С | 0.049 | 0.050 | 0.059 | 0.074 | 0.089 | 0.079 | 0.046 | 0.053 |
| X–Y | 0.049 | 0.052 | 0.045 | 0.031 | 0.050 | 0.046 | 0.035 | 0.071 | |
| ∇2ρ(r) | С–С | +0.047 | +0.045 | +0.040 | +0.024 | +0.120 | +0.111 | +0.048 | +0.045 |
| X–Y | +0.047 | +0.044 | +0.044 | +0.041 | +0.003 | +0.019 | +0.052 | +0.084 | |
| he(r) | С–С | –0.006 | –0.009 | –0.012 | –0.017 | –0.030 | –0.024 | –0.007 | –0.010 |
| X–Y | –0.006 | –0.009 | –0.007 | –0.001 | –0.002 | –0.001 | –0.003 | –0.014 | |
| EA–B, ккал/моль | С–С | 8.5 | 9.0 | 10.9 | 14.2 | 19.3 | 16.6 | 8.2 | 9.8 |
| X–Y | 8.5 | 9.3 | 7.7 | 4.7 | 10.7 | 9.4 | 5.9 | 15.4 |
Анализируя знак лапласиана электронной плотности ∇2ρ(r) в KT разрывающихся связей, мы видим, что для всех рассматриваемых ПС он положительный при отрицательном знаке локальной электронной энергии he(r). Также в этих точках для всех рассматриваемых связей выполняется соотношение 1/4∇2ρ(r) < |v(r)|. Данные взаимодействия принято относить в теории AIM к “промежуточным”, так как по своей топологии они относятся к взаимодействиям закрытых оболочек, тогда как вклады потенциальной и кинетической энергии соответствуют ковалентным связям. В то же время в исходных циклах разрывающиеся связи изначально были ковалентными. Таким образом, в результате реакции происходит переход одного типа связи в другой при образовании ПС с последующим полным разрывом связей и образованием конечных диенов.
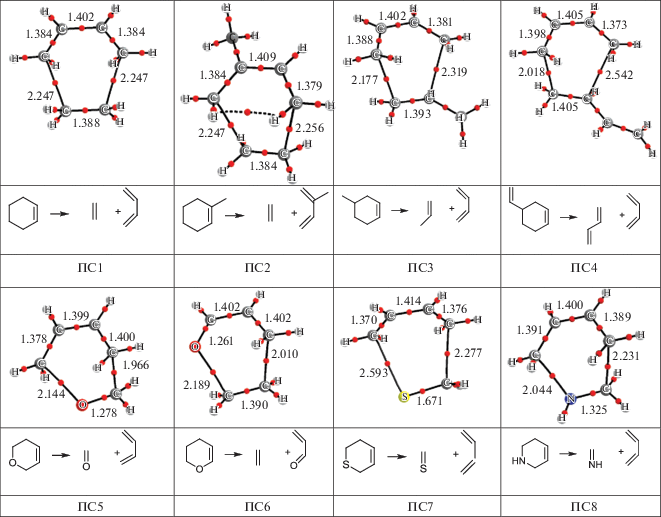
Интересно было понять, когда происходит этот переход в процессе реакции распада циклов и как на него влияет наличие заместителей в исходном цикле. Мы проанализировали, как меняется лапласиан электронной плотности в KT разрывающихся связей вдоль координаты реакции. Для этого были изучены с помощью QTAIM промежуточные структуры, полученные из процедуры IRC. Нас интересовало, как будет изменяться знак лапласиана электронной плотности в КТ разрывающихся связей вдоль координаты реакции распада циклов, так как при его изменении с отрицательного на положительный происходит изменение характера связи (рис. 2).
Рис. 2.
Зависимости лапласиана электронной плотности от координаты реакции для двух рвущихся связей: С–С (1), X–Y (2).
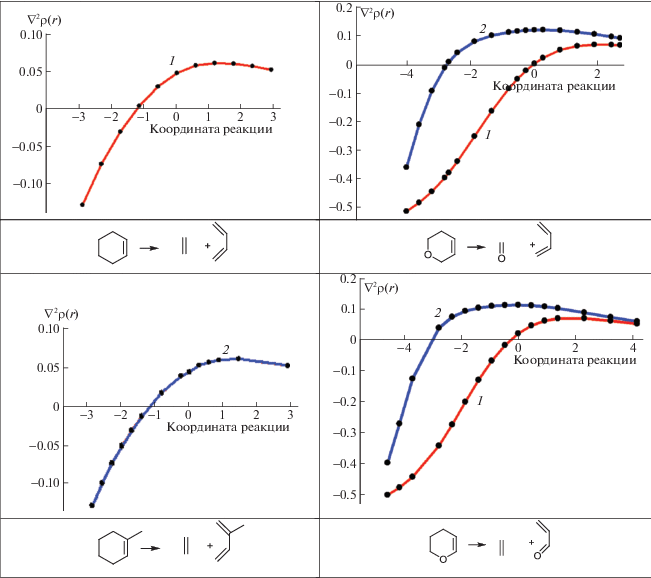
Рис. 2.
Окончание
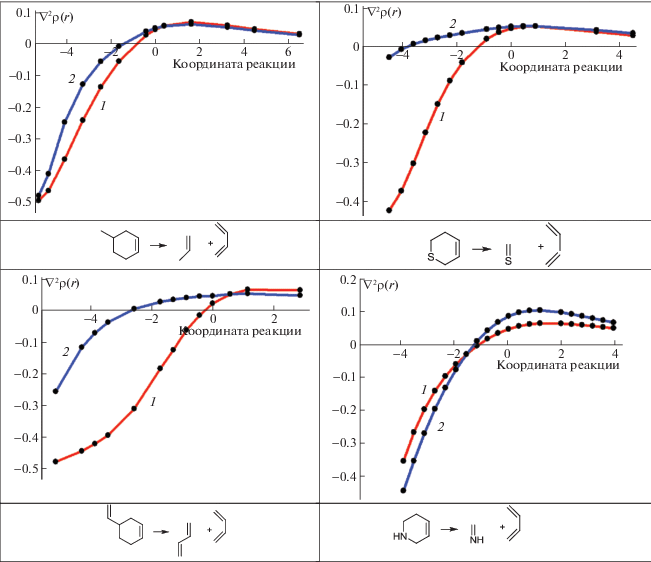
Для циклогексена мы видим полностью синхронное изменение ∇2ρ(r) вдоль координаты реакции, т.е. разрыв связей происходит одновременно, что подтверждает концертный механизм реакции через шестичленное ПС. Интересно отметить, что изменение знака лапласиана происходит не непосредственно в ПС (координата реакции 0), а несколько раньше. Характер кривой свидетельствует о плавном, а не скачкообразном перетекании электронной плотности. После прохождения переходного состояния уменьшение значения |∇2ρ(r)| происходит очень медленно, т.е. между продуктами продолжают сохраняться слабые межмолекулярные взаимодействия. Критические точки (3, –1) имеются вплоть до расстояния С–С 5 Å, что можно видеть, анализируя зависимости ∇2ρ(r) от длины разрывающейся связи (рис. 3).
Рис. 3.
Зависимость лапласиана электронной плотности от длины С−С-связи для реакции распада циклогексена.
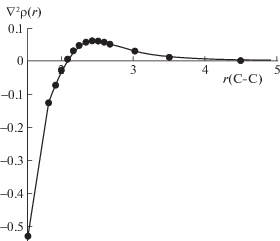
По данному графику можно определить и расстояние, при котором происходит изменение характера связи С–С, оно составляет 2.078 Å для реакции распада циклогексена.
Как было отмечено ранее, представляло интерес оценить влияние заместителей и гетероатомов в циклогексеновом кольце на прохождение реакции согласованного распада циклов. Это важно для правильного выбора рвущейся и образующейся связей для М3ПП, так как заместители, скорее всего, будут оказывать влияние на прочность разрывающихся связей, следовательно, реакция будет идти не синхронно, и необходимо оценить, в каком порядке они будут трансформироваться.
Как видно из рис. 2, в случае метилциклогексена, когда метильный заместитель находится у двойной связи, и продуктами реакции являются этилен и изопрен, никаких изменений в характере кривой не происходит (рис. 2) и значение ∇2ρ(r) обеих С–С-связей вдоль координаты реакции меняется так же, как и в случае циклогексена и, таким образом, метильный заместитель при двойной связи никак не должен повлиять на параметры реакции распада замещенного циклогексена. Совсем иная картина наблюдается для метилциклогексена, когда метильный заместитель находится в β-положении к двойной связи, а продуктами реакции являются бутадиен и пропен (рис. 2). Зависимости для связей уже не совпадают, как в двух предыдущих случаях; С–С(СН3)-связь перестает быть ковалентной несколько раньше. Это следует и из геометрии переходного состояния, так как эта связь длиннее (рис. 1). В то же время другая С–С-связь без метильного заместителя трансформируется точно так же, как в циклогексене. После прохождения энергетического барьера дальнейший распад происходит синхронно для обеих связей.
Совсем другая картина наблюдается в случае реакции распада винил-циклогексена, когда винильный заместитель находится в β-положении по отношению к двойной связи, и продуктами реакций являются две молекулы бутадиена. Во-первых, связь С–С(СН=СН2) очень слабая, т.е. мы можем видеть, как винильный заместитель ослабил связь в цикле. Скорее всего, с этим связана ранняя трансформация данной связи, задолго до достижения системой ПС. Обращает на себя внимание также изменение характера кривой оставшейся С–С-связи – она меняет свою форму по сравнению с таковой для связей в циклогексене и метилциклогексенах, и трансформация связи происходит ближе к переходному состоянию.
Большое влияние на путь реакции распада оказывает гетероатом в цикле. Так, для 6-дигидро-2H-пирана (С5H9O) изменение знака лапласиана для С–О-связи происходит значительно раньше, чем для С–С (рис. 2), и весь процесс будет определяться поведением С–С-связи, так как С–O-связь задолго до подхода к переходному состоянию уже перешла из ковалентной в промежуточную. В этом случае опять для другой С–С-связи изменение знака лапласиана происходит позже, практически в переходном состоянии. Интересно отметить, что для 3,4-дигидро-2H-пирана, когда атом кислорода находится в α-положении по отношению к двойной связи, и трансформации, как и в предыдущем случае, подвергается С–О-связь, имеем полностью идентичный график.
При распаде 3,6-дигидро-2H-тиапирана наблюдаются серьезные отличия в трансформации связей по сравнению с 6-дигидро-2H-пираном (т.е. при переходе от кислорода к сере в цикле). Во-первых, график изменения характеристик С–С-связи выглядит снова как в случае циклогексена; во-вторых, C–S-связь очень рано меняет свой характер, более того, согласно графику, она и была достаточно слабой, т.е. и в этом случае прохождение реакции определяется C–C-связью. Самый необычный вид имеет зависимость лапласиана электронной плотности в КТ разрывающихся связей для 1,2,3,6-тетрагидропиридина (С5H9N). Как видно из рис. 2, изначально связь С–N в данном соединении прочнее С–С-связи, что, вероятно, вызвано дополнительным p–σ-взаимодействием между неподеленной электронной парой атома азота и СH2-фрагментом. Поэтому она меняет свой характер практически одновременно с С–С-связью, для которой зависимость лапласиана электронной плотности в КТ от координаты реакции не отличается от таковой для циклогексена.
Таким образом, анализируя изменение знака лапласиана вдоль координаты реакции распада циклов, можно сделать вывод, что наличие заместителя или гетероатома в цикле может нарушать синхронность концертного механизма реакции. В случае алкильных заместителей значение имеет не только тип заместителя, но и его положение в кольце. Для гетероциклов важен тип разрывающейся связи, т.е. какой гетероатом входит в кольцо. Однако, в любом случае прохождение через потенциальный барьер лимитируется разрывом С–С-связи. Полученные результаты позволили корректно выбрать модель метода пересекающихся парабол (М3ПП) и провести расчеты с ее использованием [19, 21]. В табл. 2 приводены значения энергии активации, полученные М3ПП в сравнении с результатами квантово-химических расчетов (B3LYP/6-311++G**).
Таблица 2.
Энергии активации, полученные M3ПП и DFT (ккал/моль)
| М3ПП | B3LYP/6-311++G** | |
|---|---|---|
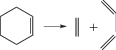 |
62.9 | 64.4 |
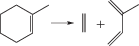 |
64.5 | 62.8 |
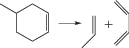 |
62.4 | 63.7 |
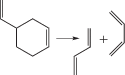 |
59.8 | 52.4 |
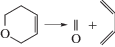 |
50.0 | 52.3 |
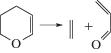 |
51.2 | 54.0 |
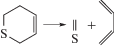 |
– | 42.2 |
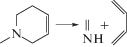 |
54.1 | 49.4 |
Как видно из табл. 2, данные двух методов вполне удовлетворительно согласуются друг с другом для всех реакций.
ЗАКЛЮЧЕНИЕ
С помощью QTAIM проведен топологический анализ ПС-реакций распада циклов различного строения. Изучена зависимость лапласиана электронной плотности в критических точках разрывающихся связей от координаты реакции вблизи переходного состояния. Показано влияние алкильного заместителя и гетероатома в цикле как на геометрию ПС, так и на путь реакции. Установлено, что метильный заместитель и атом азота не оказывают значительного воздействия, в отличие от реакций распада гетероциклов, содержащих атом кислорода, серы, а также винильный заместитель. В то же время во всех случаях лимитирующей является трансформация С–С-связи в гетероциклах и С–С-связи, не затронутой воздействием заместителя для винилциклогексена. Полученные результаты позволили корректно выбрать модель метода пересекающихся парабол – М3ПП. Рассчитанные методами М3ПП и DTF энергии активации реакции показали хорошее согласие между собой.
Работа выполнена по теме государственного задания № 0089-2019-0004.
Список литературы
Ингольд С.К. Теоретические основы органической химии. М.: Мир, 1973, 1056 с.
Denisov E.T., Sarkisov O.M., Likhtenshtein G.I. Chemical Kinetcs: Fundamentals and New Developments. Amsterdam: Elsevier, 2003, p. 268.
Семенов Н.Н. Избранные труды. Т. 3. О некоторых проблемах химической кинетики и реакционной способности. М.: Наука, 2005. С. 37.
Сергеев Г.Б., Смирнов В.В. Молекулярное галогенирование олефинов. М.: Изд-во МГУ, 1985. 240 с.
Tsang W., Rosado-Reyes C.M. // J. Phys. Chem. A. 2015. V. 119. P. 7155.
Tsang W. // J. Phys. Chem. 1986. V. 90. P. 414.
Simmie J.M. // Int. J. Chem. Kinet. 1978. V. 10. P. 227.
Huybrechts G., Luyckx L., Vandenboom Th., Van Mele B. // Int. J. Chem. Kinet. 1977. V. 9. P. 283.
Walsh R., Wells J.M. // J. Chem. Soc. Perkin Trans. 2. 1976. V. 52. P. 52.
Herndon W.C., Grayson C.R., Manion J.M. // J. Org. Chem. 1967. V. 32. P. 526.
Frey H.M., Lodge S.P. // J. Chem. Soc. Perkin Trans. 2. 1979. P. 1463.
Besseris G.J., Kiefer J.H., Zhang Q., Walker J.A. et al. // Int. J. Chem. Kinet.1995. V. 27. P. 691.
Sidhu S.S., Kiefer J.H., Lifshitz A., Tamburu C. et al. // Int. J. Chem. Kinet. 1991. V. 23. P. 215.
Покидова Т.С., Денисов Е.Т., Шестаков А.Ф. // Нефтехимия. 2009. Т. 49. С. 343.
Емельянова Н.С., Покидова Т.С. // Изв. АН. Сер. хим. 2016. № 10. С. 2333.
Денисов Е.Т., Покидова Т.С. // Нефтехимия. 2018. Т. 58. С. 113.
Покидова Т.С., Шестаков А.Ф. // Журн. физ. химии. 2009. V. 83. P. 2049.
Денисов Е.Т., Покидова Т.С. // Успехи химии. 2012. Т. 81. С. 415.
Покидова Т.С., Емельянова Н.С. // Изв. АН. Сер. хим. 2018. Т. 67. С. 1558.
Denisov E.T., Denisova T.G., Pokidova T.S. Handbook of Free Radical Initiators. Hoboken, New Jersey: Wiley. 2003. 879 p.
Денисов Е.Т. // Кинетика и катализ. 2008. Т. 49. С. 333.
Покидова Т.С., Денисов Е.Т. // Кинетика и катализ. 2001. V. 42. P. 805.
Bladow L.L., Stopera C.J., Thweatt W.D., Page M. // J. Phys. Chem. A. 2010. V. 114. P. 4304.
Бэйдер Р. Атомы в молекулах. Квантовая теория. М.: Мир, 2001, 532 с.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E. et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford (CT), 2013.
Todd A., Keith T.K. AIMAll (Version 15.05.18). Gristmill Software, Overland Park KS, USA, 2015 (aim.tkgristmill.com).
Espinosa E., Molins E., Lecomte C. // Chem. Phys. Lett. 1998. V. 285. P. 170.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии