

Журнал физической химии, 2020, T. 94, № 5, стр. 719-725
Моделирование таутомерного равновесия и спектра поглощения 4,5-диметил-2-(2'-гидроксифенил)имидазола
Д. П. Капуста a, *, А. М. Кулакова a, М. Г. Хренова a, b
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Российская академия наук, Институт биохимии им. А.Н. Баха, ФИЦ Биотехнологии
Москва, Россия
* E-mail: limaka@mail.ru
Поступила в редакцию 17.07.2019
После доработки 17.07.2019
Принята к публикации 17.09.2019
Аннотация
На примере таутомерных превращений 4,5-диметил-2(2'-гидроксифенил)имидазола продемонстрированы возможности нового интерфейса для расчетов методом молекулярной динамики с потенциалами комбинированного метода квантовой механики / молекулярной механики. В полученных траекториях наблюдается таутомеризация органической молекулы в водном растворе, а также формирование сетки водородных связей, позволяющей осуществить внутримолекулярный перенос протона между дистанцированными атомами. Расчеты полос поглощения таутомерных форм позволили провести интерпретацию экспериментального спектра поглощения для рассматриваемого соединения.
Таутомеризация органических соединений – внутримолекулярный перенос протона, сопровождающийся изменением порядка связей в молекуле [1, 2]. Эти процессы крайне чувствительны к составу растворителя, его полярности, способности образовывать водородные связи, величине pH [1, 2]. Моделирование таких процессов является сложной задачей, так как требует адекватного описания растворителя, особенно когда речь идет о воде. Простейшие континуальные модели не описывают явно водородные связи, а также другие специфические взаимодействия [3]. Описание молекул воды в рамках классических силовых полей более предпочтительно. Однако такое описание не подходит, если в переносе протона должны участвовать молекулы растворителя, поскольку классические силовые поля не описывают разрыв или образование связей. Наиболее универсальный в данном случае подход – описание молекулы растворенного вещества и молекул растворителя методами квантовой химии.
Другой особенностью моделирования процессов в растворе является то, что молекулы растворителя достаточно лабильные и одна или несколько структур, соответствующих минимумам на поверхности потенциальной энергии, не могут в полной мере описать весь конформационный набор состояний. Это приводит к необходимости использовать метод молекулярной динамики (МД) для наиболее реалистичного описания системы [4, 5].
При построении модели небольшой органической молекулы в ячейке из молекул воды размер системы составляет около 1000 атомов. Такие системы практически невозможно рассчитывать при использовании гибридных функционалов метода функционала электронной плотности (DFT) с двухэкспонентными базисами с поляризационными функциями, которые наиболее часто используются для описания квантовых подсистем на высоком уровне точности [6]. Альтернативой является применение менее точных расчетов с обобщенно-градиентными функционалами и комбинированными базисами гауссовых функций и плоских волн [7]. Однако ухудшение квантово-химического протокола может приводить к появлению артефактных результатов. Более перспективным является применение комбинированного метода квантовой механики/молекулярной механики (КМ/ММ) [8, 9], выделяя в квантово-механическую (КМ) подсистему только органическую молекулу и ближайшие молекулы растворителя, при этом всю остальную систему описывая в рамках классических силовых полей.
В рамках данной работы мы разработали новый интерфейс метода КМ/ММ для совместного применения программного пакета Firefly [10] (частично основан на GAMESSUS [11]) и NAMD [12]. В качестве модельной системы использовался водный раствор 4,5-диметил-2(2'-гидроксифенил)имидазола (ДМГИ) в нейтральной среде. По результатам работы показана возможность наблюдать внутримолекулярный перенос протона без участия молекул растворителя и выстраивание цепочки водородных связей между двумя дистанцированными атомами, участвующими в таутомеризации. Также были проведены расчеты вертикальных энергий электронного перехода S0–S1 для набора кадров из молекулярно-динамической траектории и продемонстрирована возможность описания уширения спектральной полосы за счет конформационной динамики системы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы
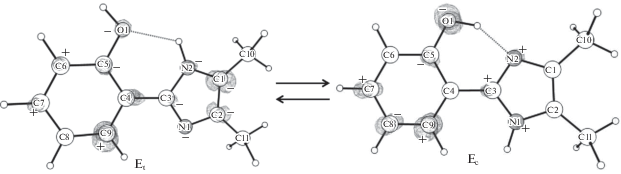
В работе рассматриваются таутомерные формы нейтральной молекулы 4,5-диметил-2-(2'-гидроксифенил)имидазола в водном растворе. Согласно литературным данным, в воде при pH 8.4 присутствуют три таутомерных формы: цис-енольная (Ec), транс-енольная (Et) и кето- (K) формы (рис. 1) [13]. При построении модельной системы первоначально проводилась оптимизация геометрических параметров ДМГИ методом функционала электронной плотности в варианте PBE0/6-31G**. Этот протокол хорошо описывает равновесные геометрические параметры, а также относительные энергии конформеров молекул [6], в том числе органических хромофоров, стабилизированных водородными связями [14]. Расчеты всех систем проводились в основном синглетном состоянии, S0.
Рис. 1.
Таутомерные формы нейтральной молекулы 4,5-диметил-2-(2'-гидроксифенил)имидазола: цис-енольная (Ec), транс-енольная (Et) и кето- (K).

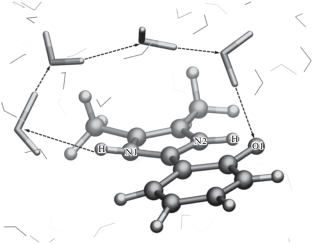
Далее молекулу ДМГИ помещали в прямоугольный параллелепипед из молекул воды так, чтобы расстояние от органической молекулы до границ ячейки составляло не менее 10 Å. Размер модельной системы составил 1394 атома в ячейке размером 30 × 30 × 30 Å3 (рис. 2).
Рис. 2.
Модельная система для расчетов методом КМ/ММ. Здесь и далее шаростержневое представление относится к ДМГИ, стержнями показаны молекулы воды в КМ-подсистеме, линиями – молекулы воды в ММ-подсистеме. Атомы азота и кислорода, участвующие в таутомеризации маркированы O1, N1 и N2. Пунктирной окружностью выделен атом, от которого рассчитывается расстояние до молекул воды при включении их в КМ- или ММ-подсистему.

Для первичного уравновешивания модельной системы проводились расчеты классической молекулярно-динамической траектории длиной 50 пс. Классические и КМ/ММ МД-расчеты проводились в NPT-ансамбле, при температуре 300 К и давлении 1 атм с использованием термостата Ланжевена с параметром связывания 50 фс–1 и баростата Нозе–Гувера [15], шаг интегрирования составлял 0.5 фс. Взаимодействия рассчитывались для атомов, находящихся на расстоянии менее чем 12 Å в классическом расчете и для ММ-подсистемы в КМ/ММ расчете. В ходе классической молекулярной динамики ДМГИ описывался набором параметров силового поля CGenFF [16], а молекулы воды – параметрами TIP3P [17]. Эти расчеты проводились в программном пакете NAMD 2.12 [12]. Классические силовые поля не позволяют описывать таутомерные переходы, то есть акты образования и разрыва химических связей, поэтому все последующие расчеты проводились методом молекулярной динамики с потенциалами КМ/ММ. Здесь стоит отметить особенность рассматриваемых систем: молекулы воды очень лабильные и в ходе расчета траектории могут входить в ближайшее окружение ДМГИ и выходить из него. Иными словами, если в начале моделирования жестко фиксировать атомы, которые будут входить в КМ-подсистему, то в ходе молекулярно-динамического расчета КМ- и ММ-молекулы воды перемешаются, что повлияет на описание взаимодействий между растворенной молекулой и растворителем. Поэтому в данной работе за основу был взят алгоритм, разработанный авторами пакета NAMD для проведения КМ/ММ МД-расчетов, позволяющий переопределять КМ-подсистему в ходе расчетов, руководствуясь определенными правилами [18]. Интерфейс, предложенный разработчиками, предполагал взаимодействие квантово-механического пакета ORCA [19] c МД-пакетом NAMD [12]. В нашей работе мы создали новый интерфейс, позволяющий проводить расчеты методом КМ/ММ МД, используя для расчета энергий и сил в КМ-подсистеме программный пакет Firefly [10]. Это открывает ряд новых возможностей, в частности, эффективные высокопараллельные расчеты на суперкомпьютерах, а также применение всего арсенала многоконфигурационных методов и наиболее точных на сегодняшний день вариантов теории возмущения, реализованных в Firefly [20].
При проведении КМ/ММ МД-расчетов КМ-подсистема включала в себя молекулу ДМГИ и молекулы воды, у которых хотя бы один из атомов оказывался на расстоянии 7 Å от атома углерода, находящегося между атомами азота в имидазольном кольце (рис. 2). Такое условие позволило включать только ближайшие к ДМГИ молекулы воды, которые образуют водородные связи с атомами азота и кислорода, участвующими в процессе таутомеризации, а также формировать цепочку водородных связей для переноса протона между дистанцированными атомами. Комбинация NAMD/Firefly позволяет на каждом шаге МД-траектории обновлять состав молекул растворителя в КМ-подсистеме, поэтому состав растворителя менялся при переходе от кадра к кадру в зависимости от расстояния до растворенной молекулы.
Молекулярно-динамические расчеты начинались с трех модельных систем, находящихся в различных таутомерных формах, длина каждой из траекторий составила 4 пс. Расчеты проводились методом PBE0/6-31G**/TIP3P. Дополнительно получена молекулярно-динамическая траектория большей длины в минимальном базисе 3‑21G. Это было необходимо для наблюдения заформированием сетки водородных связей между атомами N1 и O1, расположенными с противоположных сторон хромофора. Выбранный вариант КМ/ММ позволяет учитывать электростатические взаимодействия КМ- и ММ-подсистем. Для КМ-подсистемы вклады от ММ-атомов учитываются за счет вкладов от точечных зарядов, предписанных силовым полем, в одноэлекторнную часть гамильтониана квантовой подсистемы. Нековалентные взаимодействия на ММ-уровне учитываются за счет электростатического члена, рассчитывающегося из зарядов ММ-атомов и зарядов Малликена атомов КМ-подсистемы, а также ван-дер-ваальсовых взаимодействий, параметры для которых для всех атомов системы берутся из силового поля [16].
Для описания спектров поглощения всех таутомерных форм использовались молекулярные кластеры, полученные путем выделения из различных кадров молекулярно-динамической траектории молекулы ДМГИ вместе с первой сольватной оболочкой. Энергии вертикальных переходов в первое возбужденное синглетное состояние рассчитывались нестационарным методом функционала электронной плотности (TDDFT) c функционалом ωB97x-D3 в базисе def2-SVP в программном пакете ORCA [19]. Такой подход удовлетворительно показал себя при описании S0–S1-переходов для органических хромофоров [21–23]. Известно, что метод TDDFT дает завышенные энергии электронных переходов, поэтому обычной практикой является добавление аддитивного члена для лучшего согласия расчетов и эксперимента [24]. В нашем случае мы вычитали 1000 см–1 из всех получаемых в расчете значений. Для каждой из таутомерных форм расчеты энергий вертикальных переходов проводились в 50 точках траектории.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Расчеты методом молекулярной динамики с КМ/ММ потенциалами позволяют наблюдать акты образования и разрыва химических связей, в нашем случае таутомерные превращения. На рис. 3 показан пример переноса протона от атома N2 на атом O1 в ходе расчета (здесь и далее названия атомов соответствуют рис. 3). В качестве исходной структуры использовалась кето-форма, K, протонированная по двум атомам азота. В ходе молекулярно-динамического расчета протон переходит на атом кислорода O1 с образованием цис-енольной формы, Ec.
Рис. 3.
Распределение расстояний O1···H (серым) и N2···Н (черным) вдоль молекулярно-динамической траектории, демонстрирующее таутомерный переход между цис-енольной и кето-формами в ходе молекулярно-динамического моделирования.
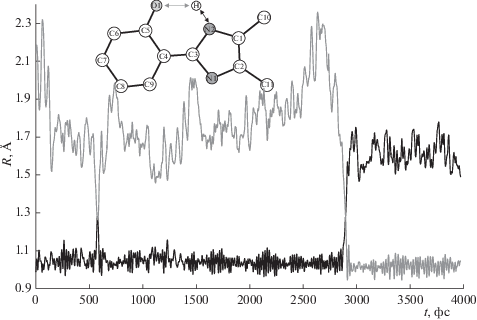
Важно отметить значимость учета водного окружения при моделировании системы. Согласно экспериментальным данным по спектрам поглощения [13], которые будут подтверждены расчетами энергий вертикальных электронных переходов далее в тексте, кето-форма преобладает в равновесии над нейтральными формами ДМГИ. Тестовые расчеты ДМГИ в газовой фазе показали невозможность локализовать минимум на поверхности потенциальной энергии, соответствующий кето-форме K. Добавление двух молекул воды, образующих водородные связи с атомами O1 и N1 органической молекулы, также не позволили локализовать минимум, соответствующий форме K.
Другое обстоятельство связано с корректной оценкой относительных энергий конформеров. Согласно нашим расчетам в газовой фазе, форма Et на 7 ккал/моль выше по энергии, чем Ec, что соответствует константе равновесия порядка 10–5 при комнатной температуре и 10–16 при 100 K. Добавление только двух молекул воды снижает разность энергий до 3 ккал/моль, что уже соответствует более реалистичным значениям. Дальнейшее расширение модельной системы приводит к появлению множества конформаций молекул растворителя, и становится невозможным сравнивать относительные энергии таутомеров. В таких случаях принято использовать метод молекулярной динамики с ограничивающим потенциалом, позволяющий проводить серию расчетов, в которых сканируются области конформационного пространства вблизи заданного значения координаты реакции [25]. Эти расчеты являются крайне ресурсоемкими, поэтому в данной работе мы изучали принципиальную возможность образования сетки водородных связей для переноса протона между атомами N1 и O1. Для этого мы проводили более продолжительный расчет, длина траектории составила 40 пс, и использовали менее затратный метод описания КМ-подсистемы – PBE0/3-21G. На рис. 4 показана цепочка из молекул воды, позволяющая осуществить перенос протона с атома N1 на атом O1. Она образуется через 15 пс после начала расчета и далее остается в основном (около 80% времени) стабильной в ходе траектории. Эти результаты могут быть использованы в качестве стартового приближения для поиска координаты реакции переноса протона.
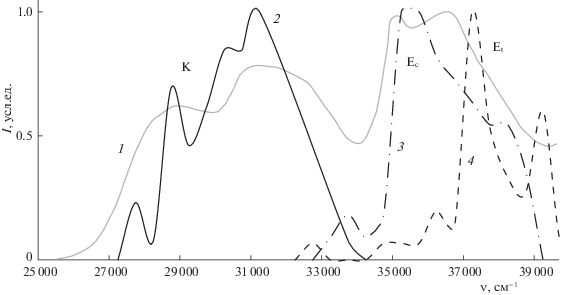
В работе [13] представлены данные электронных спектров для молекулы ДМГИ в водном растворе при pH 8.4, однако соотнесение экспериментальных полос поглощения с таутомерными формами без проведения расчетов является затруднительным. Мы использовали три молекулярно-динамические траектории с различными таутомерами ДМГИ и случайным образом выбирали по 50 кадров. Системы редуцировались так, чтобы вокруг растворенной молекулы была одна сольватная оболочка, и проводили расчет энергии вертикального S0–S1-перехода. Далее проводилась статистическая обработка результатов. Полученные значения кластеризовались с шагом 500 см–1, после чего строились распределения по волновым числам. После чего распределения смещались на 1000 см–1, чтобы добиться лучшего согласия с экспериментом. На рис. 5 представлено наложение экспериментального спектра, полученного оцифровкой спектра поглощения в водном растворе при pH 8.4 на рис. 1б в работе [13] и рассчитанных спектров для каждой из таутомерных форм. Обе енольные формы характеризуются полосами поглощения в области 33 000–39 000 см–1. Для кето-формы характерны меньшие значения волновых чисел в области 27 000–32 000 см–1. Наложение рассчитанных и экспериментальных спектров позволяет провести отнесение полос соответствующим таутомерным формам. Максимум полосы поглощения около 35 000 см–1 относится к цис-енольной форме, а полоса с максимумом в районе 36 500 см–1 – к транс-енольной. Таким образом, два максимума широкой полосы в этой области соответствуют двум таутомерам. Согласно данным расчетов, широкую полосу с двумя максимумами в области меньших волновых чисел стоит отнести к поглощению кето-формы, а наличие двух максимумов можно объяснить двумя популяциями несколько отличающихся друг от друга конформаций.
Рис. 5.
Экспериментальный спектр поглощения ДМГИ в водном растворе при pH 8.4 (серая линия, 1) [13] и рассчитанные полосы поглощения для кето- (черная сплошная линия, 2), цис-енольной (черная штрихпунктирная линия, 3) и транс-енольной (черная пунктирная линия, 4) форм.
Чтобы объяснить относительные положения максимумов полос поглощения цис- и транс-енольных форм, мы провели дополнительные расчеты в газовой фазе. В рамках тех же вычислительных протоколов рассчитаны энергии вертикальных электронных переходов и разностные электронные плотности между основным и первым возбужденным электронным состояниями. По результатам расчетов энергия вертикального S0,min–S1-перехода составила 37 743 см–1 для Eс-формы и 39 398 см–1 для Et-формы. Относительное расположение энергий вертикальных электронных переходов в газовой фазе воспроизводит таковое для рассчитанных полос поглощения в растворе (рис. 5). Проанализируем карты разностной электронной плотности (рис. 6). Для обеих систем характерно уменьшение электронной плотности на атоме кислорода O1 при переходе в S1-состояние. В случае транс-конформера присутствует сильная внутримолекулярная водородная связь N2–H···O1, которая стабилизирует основное состояние и дестабилизирует возбужденное; в цис-конформере такой связи нет. Это и приводит к синему сдвигу Et – относительно Ec-формы.
Рис. 6.
Разностная электронная плотность (S1–S0) для Ec- и Et-форм. Плюсами показаны области увеличения электронной плотности при переходе из S0- в S1-состояние, минусами – области уменьшения.
Таким образом, применение метода молекулярной динамики с КМ/ММ потенциалами позволило описать переходы между таутомерными формами нейтральной молекулы ДМГИ в водном растворе. На основании рассчитанных спектров поглощения проведена интерпретация экспериментального спектра поглощения и проведено отнесение полос. Анализ разностных электронных плотностей при переходе из основного в первое возбужденное электронное состояние позволило объяснить синий сдвиг транс-енольной формы относительно цис-енольной формы ДМГИ.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 18-03-00605. Работа выполнена с использованием оборудования Центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ имени М.В. Ломоносова.
Список литературы
Antonov L. Tautomerism: Concepts and Applications in Science and Technology. Wiley, 2016. https://doi.org/10.1002/9783527695713
Antonov L. (ed.). Tautomerism: Methods and Theories. Wiley, 2013. https://doi.org/10.1002/9783527658824
Mennucci B., Cammi R. (ed.) Continuum solvation models in chemical physics: from theory to applications. Wiley, 2008,
Fedorov D.G., Sugita Y., Choi C.H. // J. Phys. Chem. B. 2013. V. 117. № 26. P. 7996. https://doi.org/10.1021/jp4029529
Khrenova M.G., Kapusta D.P., Babchuk I.V., Meteleshko Y.I. // J. Supercomput. 2018. V. 5. № 3. P. 70. https://doi.org/10.14529/jsfi180312
Mardirossian N., Head-Gordon M. // Mol. Phys. 2017. V. 115. № 19. P. 2315. https://doi.org/10.1080/00268976.2017.1333644
Vande Vondele J., Krack M., Mohamed F. et al. // Comput. Phys. Commun. 2005. V. 167. № 2. P. 103. https://doi.org/10.1016/j.cpc.2004.12.014
Warshel A., Levitt M. // J. Mol. Biol. 1976. V. 103. № 2. P. 227. https://doi.org/10.1016/0022-2836(76)90311-9
Senn H.M., Thiel W. // Angew. Chem. 2009. V. 48. № 7. P. 1198. https://doi.org/10.1002/anie.200802019
Granovsky A.A. Firefly version 8.0.0 // URL: http://classic.chem.msu.su/gran/firefly/index.html. 2014.
Schmidt M.W., Baldridge K.K., Boatz J.A. // J. Comput. Chem. 1993. V. 14. № 11. P. 1347. https://doi.org/10.1002/jcc.540141112
Phillips J.C., Braun R., Wang W. et al. // J. Comput. Chem. 2005. V. 26. № 16. P. 1781. https://doi.org/10.1002/jcc.20289
Bräuer M., Mosquera M., Pérez-Lustres J.L., Rodríguez-Prieto F. // J. Phys. Chem. A. 1998. V. 102. № 52. P. 10736. https://doi.org/10.1021/jp983291v
Nemukhin A.V., Grigorenko B.L., Khrenova M.G., Krylov A.I. // J. Phys. Chem. B. 2019. https://doi.org/10.1021/acs.jpcb.9b00591
Martyna G.J., Tobias D.J., Klein M.L. // J. Chem. Phys. 1994. V. 101. № 5. P. 4177. https://doi.org/10.1063/1.467468
Vanommeslaeghe K., Hatcher E., Acharya C. et al. // J. Comput. Chem. 2010. V. 31. № 4. P. 671. https://doi.org/10.1002/jcc.21367
Jorgensen W.L., Chandrasekhar J., Madura J.D. et al. // J. Chem. Phys. 1983. V. 79. № 2. P. 926. https://doi.org/10.1063/1.445869
Melo M.C., Bernardi R.C., Rudack T. et al. // Nat. Methods. 2018. V. 15. № 5. P. 351. https://doi.org/10.1038/nmeth.4638
Neese F. // Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012. V. 2. № 1. P. 73. https://doi.org/10.1002/wcms.81
Granovsky A.A. // J. Chem. Phys. 2011. V. 134. № 21. P. 214113. https://doi.org/10.1063/1.3596699
Meteleshko Y.I., Nemukhin A.V., Khrenova M.G. // Photochem. Photobiol. Sci. 2019. V. 18. № 1. P. 177. https://doi.org/10.1039/c8pp00361k
Khrenova M.G., Meteleshko Y.I., Nemukhin A.V. // J. Phys. Chem. B. 2017. V. 121. № 43. P. 10018. https://doi.org/10.1021/acs.jpcb.7b07533
Khrenova M.G., Polyakov I.V., Grigorenko B.L. et al. // Ibid. 2017. V. 121. № 47. P. 10602. https://doi.org/10.1021/acs.jpcb.7b07517
Syzgantseva O.A., Tognetti V., Joubert L. et al. // J. Phys. Chem. A. 2012. V. 116. № 33. P. 8634. https://doi.org/10.1021/jp305269y
E. Torrie G.M., Valleau J.P. // J. Comput. Phys. 1977. V. 23. P. 187. https://doi.org/10.1016/0021-9991(77)90121-8
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии