

Журнал физической химии, 2020, T. 94, № 5, стр. 726-730
Квантово-химическое моделирование спектральных свойств красителя BODIPY с разными заместителями
a Российская академия наук, Институт проблем химической физики
Московская область, Черноголовка, Россия
b Московский физико-технический институт (государственный университет)
Долгопрудный, Россия
* E-mail: nevidimovsasha@yandex.ru
Поступила в редакцию 18.07.2019
После доработки 18.07.2019
Принята к публикации 17.09.2019
Аннотация
В данной работе методом TD-DFT изучали влияние сильных электронодонорных заместителей (3- и 4-аминофенильных групп) в положениях 3 и 5 красителя 4,4-дифтор-4-бора-3а,4а-диаза-s-индацена (BODIPY) на его спектральные свойства. Показано, что наличие аминогрупп в положении 4 фенильного кольца существенно больше, чем в положении 3 изменяет энергетические уровни вблизи высшей заполненной молекулярной орбитали, уменьшая зазор между ней и низшей свободной молекулярной орбиталью.
Молекулярные люминофоры (красители) представляют собой перспективные объекты как для фундаментальных исследований, так и для многочисленных практических применений [1–4]. Наиболее ценное преимущество молекулярных люминофоров состоит в том, что благодаря высоким коэффициентам поглощения света и большим квантовым выходам люминесценции они обладают чрезвычайно малыми пределами обнаружения в растворах. Здесь речь идет о предельных концентрациях, которые можно обнаружить методами оптической спектроскопии, порядка 10–10 М и ниже. Однако сама по себе возможность обнаруживать присутствие сверхмалых количеств молекул красителя в растворах была бы не так интересна с практической стороны, если бы оптические свойства красителей не менялись под воздействием различных соединений, которые уже представляют значительно больший интерес для обнаружения. Это могут быть и токсичные вещества, и ядовитые, наркотические, взрывчатые. В присутствии таких веществ спектральные свойства люминофора могут изменяться, что в сочетании с их высокими оптическими показателями делает возможным обнаруживать интересующие опасные вещества также в сверхмалых дозах. При этом важной задачей перед современной химической наукой стоит задача в подборе таких красителей, которые бы могли селективно менять свои свойства в присутствии определенных опасных веществ.
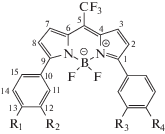
Данная работа посвящена изучению спектральных свойств аминопроизводных органического красителя BODIPY (рис. 1), про который известно, что его спектральные свойства чувствительны к присутствию различных полярных молекул [1]. Авторы [1] помещали производное рассматриваемого красителя с одной аминогруппой (соединения 2 и 3 на рис. 1) в полярную апротонную среду (1,4-диоксан, диметилсульфоксид и другие), и его люминесцентные свойства резко ухудшались. Протонирование (подкисление) полярного раствора приводило к восстановлению оптических свойств красителя. При этом краситель, не содержащий аминогрупп (соединение 1), не испытывал изменений при смене среды. Проведенные в работе [1] тяжеловесные квантово-химические расчеты методом RI–CC2 соединений 1–3 продемонстрировали принципиальную возможность изучения спектральных характеристик такого класса молекул квантово-химическими методами, а также показали, что величина энергетического зазора между высшей занятой (ВЗМО) и низшей свободной (НСМО) молекулярными орбиталями практически не зависит от наличия аминогруппы и ее расположения и составляет 6.5–7 эВ. Также в той работе было показано, что структура самих НСМО и ВЗМО не зависит от присутствия аминогрупп, а более низкой по энергии ВЗМО–1 зависит весьма существенно. В данной работе проведены квантово-химические расчеты, пусть и более скромные по вычислительной сложности, но нацеленные на более детальное изучение спектральных свойств молекул из работы [1], а также двух дополнительных производных, содержащих по две аминогруппы.
Рис. 1.
Структурные формулы пяти соединений BODIPY: 1 – все R=H, 2 – R1=NH2, 3 – R2=NH2, 4 – R1=R4=NH2, 5 – R2=R3=NH2. Не указанные R отвечают атомам Н. Нумерация атомов C, используемая в табл. 1.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для проведения квантово-химических расчетов была использована свободно распространяемая программа FireFly 8.2 [5], частично основанная на GAMESS US [6]. Расчеты проводили с привлечением оборудования центра коллективного пользования сверхвысокопроизводительными вычислительными ресурсами МГУ им. М.В. Ломоносова [7]. Сначала проводили оптимизацию геометрии молекулы 1 в небольшом базисе STO-3G в рамках ограниченного метода Хартри–Фока, поскольку стартовая молекула обладала структурой, далекой от равновесной. После этого базис был изменен на 6-311G**, и проведена дальнейшая оптимизация геометрии молекулы 1 в рамках теории функционала электронной плотности (DFT) с функционалом B3LYP5. Затем к молекуле 1 были добавлены аминогруппы в нужных местах для получения молекул 2–5 и проведена их оптимизация в том же базисе. После этого для 1–5 рассчитали матрицу вторых производных, чтобы удостовериться, что найденная точка экстремума отвечает локальному минимуму на поверхности потенциальной энергии. После подтверждения истинности минимума для всех соединений были выполнены расчеты энергии возбужденного состояния в рамках теории TD–DFT с функционалом B3LYP5. Визуализацию и анализ найденных структур и молекулярных орбиталей выполняли с помощью свободно распространяемой программы VMD [8].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Поиск оптимальной геометрии
Для всех исследуемых молекул были найдены оптимальные геометрии и доказано, что они соответствуют истинным минимумам на поверхности потенциальной энергии путем расчета матрицы вторых производных. Все найденные структуры характеризуются практически плоским ядром молекулы BODIPY. Атомы фтора в группе дифторборана развернуты перпендикулярно основной плоскости ядра BODIPY. Фенильные кольца тоже развернуты по отношению к плоскости ядра, но угол существенно меньший. Поворот фенильных колец происходит из-за стерических затруднений, как это происходит в молекуле бифенила. При этом электростатическое притяжение атома H бензольного кольца к атому F группы дифторборана делает величину этого угла близкой к 40°. Оптимальное расстояние между указанными атомами H и F составляет 0.21–0.22 нм, что соответствует длине обычной водородной связи. Поскольку атомы H бензольного кольца не способны к формированию водородных связей, здесь речь идет об обычном электростатическом притяжении двух противоположно заряженных атомов. Также небольшой вклад вносит стремление системы к π–π-сопряжению 4‑аминофенильной группы в молекулах 2 и 4 с общей плоскостью ядра BODIPY, в результате чего величина разворота этих колец уменьшается до 30–33°.
Расчет возбужденного состояния
Было также исследовано влияние аминогрупп на распределение электронной плотности в молекуле BODIPY. Для этого рассматривали заряды на атомах, вычисленные по методу Малликена для всех соединений в основном и возбужденном состояниях. В табл. 1 представлены заряды некоторых атомов C, отмеченных цифрами на рис. 1, и атомов N для соединений 1–3. Из табл. 1 видно, что в основном состоянии точечные заряды указанных атомов приблизительно одинаковы – максимальное различие достигает всего 0.03 величины элементарного электрического заряда. Формальное исключение составляют атомы C в бензольном кольце, непосредственно связанные с NH2-группой. Однако уже соседние атомы C имеют практически одинаковые заряды. Это свидетельствует о том, что в основном состоянии аминогруппа оказывает небольшое влияние на распределение электронной плотности в молекуле BODIPY. При переходе в возбужденное состояние картина кардинально меняется. Наличие аминогруппы способно изменить величину заряда атома C, к которому она прикреплена, на 0.31, а также других атомов C в самом бензольном кольце и даже в ядре BODIPY вплоть до 0.15 элементарного заряда. При этом стоит обратить внимание, что при переходе в возбужденное состояние происходит резкое увеличение заряда азота аминогруппы, а также, что в целом заряды атомов бензольного кольца, содержащего аминогруппу, становятся больше, а заряды атомов ядра BODIPY становятся меньше. Таким образом, аминогруппа существенно влияет на перераспределение зарядов в молекуле красителя, сдвигая электронную плотность от бензольного кольца к ядру.
Таблица 1.
Атомные заряды (по Малликену) для соединений 1–3 в основном и возбужденном (*) состояниях. Жирным цветом выделены атомы, заряды которых наиболее сильно изменяются при внедрении аминогруппы. Нумерацию атомов С см. на рис. 1, атомы N1 связан с С1, атом N2 – с С9, N3 – в аминогруппе
| Атом | 1 | 2 | 3 | 1* | 2* | 3* |
|---|---|---|---|---|---|---|
| B | +0.65 | +0.67 | +0.67 | +0.64 | +0.67 | +0.67 |
| N1 | –0.47 | –0.48 | –0.48 | –0.50 | –0.50 | –0.51 |
| N2 | –0.49 | –0.49 | –0.48 | –0.53 | –0.52 | –0.52 |
| C1 | +0.26 | +0.23 | +0.23 | +0.22 | +0.23 | +0.23 |
| C2 | –0.15 | –0.08 | –0.11 | –0.17 | ||
| C3 | –0.05 | –0.07 | –0.06 | –0.13 | –0.14 | –0.16 |
| C4 | +0.24 | +0.23 | +0.23 | +0.36 | +0.31 | +0.21 |
| C5 | –0.34 | –0.33 | –0.33 | –0.56 | –0.54 | –0.55 |
| C6 | +0.23 | +0.23 | +0.22 | +0.34 | +0.29 | +0.21 |
| C7 | –0.07 | –0.16 | –0.17 | –0.18 | ||
| C8 | –0.15 | –0.09 | –0.12 | –0.17 | ||
| C9 | +0.25 | +0.24 | +0.24 | +0.20 | +0.14 | +0.11 |
| C10 | –0.08 | –0.08 | –0.09 | –0.04 | +0.03 | –0.08 |
| C11 | –0.02 | –0.02 | –0.01 | –0.01 | –0.02 | +0.11 |
| C12 | –0.10 | –0.09 | +0.09 | –0.10 | –0.05 | +0.21 |
| C13 | –0.07 | +0.15 | –0.05 | –0.03 | +0.20 | +0.05 |
| C14 | –0.10 | –0.08 | –0.10 | –0.09 | –0.03 | –0.07 |
| C15 | –0.04 | –0.05 | –0.05 | –0.03 | –0.05 | +0.16 |
| N3 | –0.47 | –0.48 | –0.37 | –0.17 | ||
Рассмотрим теперь, как изменяются при этом молекулярные орбитали (табл. 2). Анализ молекулярных орбиталей показывает то же самое, что и анализ точечных зарядов: при электронном возбуждении происходит перенос электронной плотности с бензольного кольца, к которому присоединена аминогруппа, на ядро BODIPY. Величина энергетического зазора между ВЗМО и НСМО уменьшается при введении аминогруппы, причем для аминогруппы в пара-положении эффект больше. Также характерной особенностью является близкое расположение орбиталей ВЗМО, ВЗМО–1 для мета-изомера с одной NH2-группой и дополнительно ВЗМО–2 – с двумя группами. Для пара-изомера эти орбитали также отстоят на одинаковые, но заметно бóльшие расстояния. Также интересным является большая величина энергетического зазора между НСМО и НСМО+1, сравнимая с расстоянием между ВЗМО и НСМО.
Таблица 2.
Молекулярные орбитали соединений 1–5 (параметр орбиталей равен 0.2 а.е.) и их энергии (а.е.)
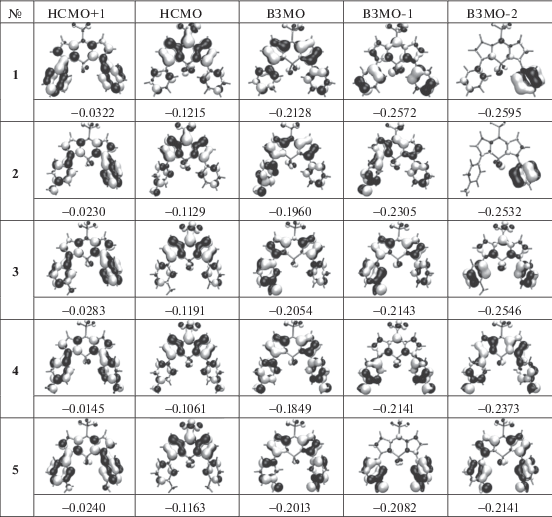
Отмеченные особенности приводят к тому, что при поглощении света заселение НСМО+1 практически не происходит. Анализ первых 20 возбужденных состояний подтверждает это. Переход ВЗМО → НСМО+1 наблюдается только при возбужденном состоянии 8 с энергией около 4.5 эВ независимо от наличия аминогрупп.
Таблица 3 показывает количественные характеристики переходов на НСМО с первых нескольких занятых орбиталей. Видно, что для мета-изомеров уже первое возбужденное состояние содержит смесь переходов с ВЗМО (вклад 65–70%) и ВЗМО–1 или ВЗМО–2 (вклад 30–35%) на НСМО. Для пара-изомеров и молекулы без NH2-группы первое возбужденное состояние на 100% состоит из перехода ВЗМО → НСМО.
Таблица 3.
Энергии электронного возбуждения (${{E}_{{v}}}$), силы осцилляторов (f) и вклады МО в возбужденные состояния молекул 1–5. В скобках показаны значения из [1]
| № | Переход | ${{E}_{{v}}}$, эВ | f | МО | % |
|---|---|---|---|---|---|
| 1 | S1 → S0 | 2.48 (2.54) | 0.634 (0.590) | ВЗ → НС | 100 (92.1) |
| S2 → S0 | 3.22 (3.53) | 0.014 (0.054) | ВЗ-2 → НС ВЗ-1 → НС |
58.0 30.7 |
|
| S3 → S0 | 3.24 (3.72) | 0.012 (0.005) | ВЗ-3 → НС ВЗ-2 → НС ВЗ-1 → НС |
56.2 30.8 11.9 (11.4) |
|
| 2 | S1 → S0 | 2.23 (2.36) | 0.613 (0.620) | ВЗ → НС | 100 (93.2) |
| S2 → S0 | 2.95 (3.25) | 0.189 (0.163) | ВЗ-1 → НС | 93.1 (74.5) | |
| S3 → S0 | 3.31 (3.57) | 0.002 (0.062) | ВЗ-2 → НС | 91.8 | |
| 3 | S1 → S0 | 2.00 (2.50) | 0.055 (0.56) | ВЗ → НС ВЗ-1 → НС |
64.5 (91.9) 35.3 |
| S2 → S0 | 2.44 (2.83) | 0.540 (0.025) | ВЗ-1 → НС ВЗ → НС |
63.5 (94.1) 35.7 |
|
| S3 → S0 | 3.24 (3.53) | 0.021 (0.054) | ВЗ-3 → НС ВЗ-2 → НС |
75.0 23.5 |
|
| 4 | S1 → S0 | 2.10 | 0.590 | ВЗ → НС | 100 |
| S2 → S0 | 2.70 | 0.239 | ВЗ-1 → НС | 97.1 | |
| S3 → S0 | 3.20 | 0.033 | ВЗ-2 → НС ВЗ-5 → НС |
73.7 23.7 |
|
| 5 | S1 → S0 | 2.00 | 0.091 | ВЗ → НС ВЗ-2 → НС |
68.5 27.3 |
| S2 → S0 | 2.05 | 0.019 | ВЗ-1 → НС | 94.7 | |
| S3 → S0 | 2.46 | 0.481 | ВЗ-2 → НС ВЗ → НС |
68.8 29.7 |
Величины энергетических переходов, показанные в табл. 3 и на рис. 2, отличаются в меньшую сторону от значений, полученных в работе [1], однако качественно согласуются с ними. Как и в [1], добавление аминогрупп приводит к уменьшению энергии, требуемой для возбуждения. Существенное отличие результатов с использованием метода расчета TD–DFT и применяемом в работе [1] методе RI–CC2 наблюдается для вклада молекулярных орбиталей мета-изомера в возбужденное состояние. Для пара-изомера и молекулы без NH2-группы данные, полученные разными методами, согласуются лучше. Это указывает на границы применимости относительно простого TD–DFT-метода по сравнению с используемым в работе [1] методом RI–CC2.
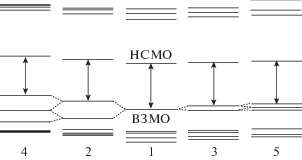
Появление новых орбиталей, близких по энергии к ВЗМО, при введении аминогруппы в мета- или пара-положения фенильного кольца и существенное изменение энергетических характеристик переходов в возбужденное состояние свидетельствует о сильном влиянии аминогруппы на электронные свойства молекул. Исходя из анализа зарядов можно предположить, что главным фактором при этом выступает неподеленная пара электронов атома N, которая взаимодействует с π-системой молекулы BODIPY. Это может отражаться в усиленном влиянии различных веществ, которые способны взаимодействовать с неподеленной парой электронов аминогруппы, таких как протон, и может приводить к значительным сдвигам в энергетических уровнях молекулы BODIPY. Причем эффект для пара-изомеров должен значительно превосходить таковой для мета-изомеров. Дополнительное исследование электронных свойств молекулы BODIPY с протонированной аминогруппой представляется интересным, но не входит в рамки настоящей работы.
Таким образом, в результате проведенной работы продемонстрированы возможности применения недорогой процедуры квантово-химического расчета методом TD–DFT с небольшим базисом 6-311G** при моделировании спектральных характеристик молекулы BODIPY и аминопроизводных. Такие расчеты позволяют качественно описать перераспределение электронной плотности при введении аминогруппы в молекулу красителя BODIPY в основном и возбужденном состояниях, которые согласуются с экспериментальными результатами и частично с данными более ресурсоемких вычислений методом RI–CC2.
В результате проведенной работы показано, что краситель BODIPY с аминофенильными заместителями способен изменять свои электронные свойства по сравнению с молекулой без аминогрупп. При этом ожидаемый эффект сильнее выражен для аминогруппы в пара-положении по отношению к ядру молекулы BODIPY, чем для мета-изомера, а если аминогрупп две, то их влияние еще более выражено.
Работа выполнена по теме гос. задания (№ 0089-2019-0003) и при финансовой поддержке Российской Федерации (№ 074-02-2018-286).
Список литературы
Petrushenko K.B., Petrushenko I.K., Petrova O.V. et al. // Asian J. Org. Chem. 2017. V. 6. P. 852.
Tomilin D.N., Petrushenko K.B., Sobenina L.N. et al. // Ibid. 2016. V. 5. P. 1288.
Товстун С.А., Иванчихина А.В., Николенко Л.М. и др. // ХВЭ. 2015. Т. 49. № 2. С. 128.
Невидимов А.В., Разумов В.Ф. // Коллоидн. журн. 2018. Т. 80. № 5. С. 556.
Firefly project: <http://classic.chem.msu.su/gran/firefly>
Schmidt M.W., Baldridge K.K., Boatz J.A. et al. // J. Comput. Chem. 1993. V. 14. P. 1347.
Воеводин В., Жуматий С., Соболев С. и др. // Открытые сист. 2012. № 7. С. 36.
Humprey W., Dalke A., Schulten K. // J. Molec. Graphics. 1996. V. 14. № 1. P. 33.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии