

Журнал физической химии, 2020, T. 94, № 5, стр. 731-737
Спектроскопические свойства молекулы сульфида бария в низколежащих электронных состояниях X1Σ+, a3Π, A'1Π, b3Σ+ и A1Σ+
А. Н. Смирнов a, В. Г. Соломоник a, *
a Ивановский государственный химико-технологический университет
Иваново, Россия
* E-mail: sol@isuct.ru
Поступила в редакцию 05.07.2019
После доработки 16.07.2019
Принята к публикации 17.09.2019
Аннотация
Методом связанных кластеров c включением однократных, двойных, тройных и четырехкратных возбуждений CCSDTQ с экстраполяцией к пределу полного базисного набора (CBS) и с учетом спин-орбитального взаимодействия вычислены спектроскопические константы re, ωe, ωexe, αe, De, βe и энергия атомизации молекулы сульфида бария BaS в основном электронном состоянии X1Σ+. Получено прекрасное согласие результатов расчетов с имеющимися экспериментальными данными: например, теоретические величины re и ωe отличаются от полученных в спектроскопических экспериментах менее чем на 0.001 Å и 1 см–1 соответственно. Показано, что корреляция 5s- и 5p-электронов атомного остова бария вносит очень большой вклад в величины молекулярных параметров. С использованием подхода, основанного на сочетании метода связанных кластеров в приближении CCSD(T)/CBS и многоисходного метода конфигурационного взаимодействия MRCISD+Q, впервые дано высокоточное теоретическое описание низколежащих возбужденных триплетных (a3Π, b3Σ+) и синглетных (A'1Π, A1Σ+) электронных состояний молекулы BaS.
Сведения о свойствах соединений щелочно-земельных металлов важны для многих отраслей науки и техники, в том числе для биохимии, материаловедения, для синтеза эффективных катализаторов и др. Наноматериалы на основе сульфидов щелочноземельных металлов, в том числе бария, находят широкое применение при производстве ряда электронных и оптических устройств [1, 2]. Между тем, имеющаяся информация о свойствах молекул этих веществ неполна, а во многих случаях и весьма неточна. Это справедливо даже для простейших двухатомных молекул сульфидов щелочноземельных металлов, в частности для молекулы сульфида бария BaS, являющейся предметом нашего исследования. Не в последнюю очередь такая ситуация объясняется не только трудностями экспериментального изучения молекул халькогенидов металлов, но и недостаточной разработанностью теоретических подходов к достаточно точному описанию свойств этих молекул.
Цели нашей работы – 1) на примере молекулы сульфида бария оценить точность описания спектроскопии и термохимии соединений тяжелых щелочноземельных элементов, которой можно достичь с применением современных методов неэмпирической квантовой химии. (Такую оценку мы проводим, сравнивая результаты ab initio расчетов характеристик основного электронного состояния X1Σ+ молекулы BaS с экспериментальными данными (спектроскопические константы основного состояния этой молекулы известны из опыта с высокой точностью [3])); 2) определить спектроскопические параметры низколежащих возбужденных электронных состояний a3Π, A'1Π, b3Σ+ и A1Σ+ молекулы BaS, сведения о которых, имеющиеся в литературе, далеко не полны.
Для решения этих задач мы разработали и применили “составной” метод: наиболее значимые вклады в величины молекулярных параметров вычисляем в самых высоких теоретических приближениях (с весьма полным учетом электронной корреляции в очень широких базисах), тогда как вклады, меньшие по величине, но требующие для своего определения проведения гораздо более трудоемких расчетов, вычисляем на менее высоких уровнях теории. Учет электронной корреляции проводим методом связанных кластеров с одной исходной конфигурацией (вплоть до уровня CCSDTQ) или/и многоисходным (с многими исходными конфигурациями) методом конфигурационного взаимодействия, MRCISD+Q. В качестве элементов применяемой нами вычислительной схемы служат эффекты неполноты базисного набора функций, эффекты корреляции остовных электронов, вклады от спин-орбитального взаимодействия, а также корреляционные вклады от высших [относительно “золотого стандарта” CCSD(T)] электронных возбуждений.
МЕТОДИКА РАСЧЕТОВ
Большинство вычислений мы провели по программе MOLPRO [4] методом связанных кластеров с учетом одно- и двукратных возбуждений и с поправкой по теории возмущений на трехкратные возбуждения, CCSD(T) [5–7], а также методом взаимодействия конфигураций, одно- и двукратно возбужденных относительно многих исходных конфигураций (MRCISD) [8, 9], с включением многоконфигурационного аналога поправки Дэвидсона на квартичные возбуждения (+Q) [10]. Метод MRCISD+Q обозначаем далее сокращением MRCI. В расчетах систем с открытой электронной оболочкой, а именно возбужденных триплетных состояний a3Π и b3Σ+ молекулы BaS и основного состояния 3P атома серы, в качестве исходной для метода CCSD(T) служила конфигурация, полученная ограниченным по спину методом Хартри–Фока для открытых оболочек (ROHF) с приведением вырожденных орбиталей к симметрично-эквивалентной форме. В методе MRCI исходной была волновая функция, получаемая многоконфигурационным методом самосогласованного поля в приближении SA-CASSCF [11, 12] в полном активном пространстве, соответствующем расселению шести электронов на четырех молекулярных орбиталях (МО), в которых преобладают 6s-орбитали атома бария и 3p-орбитали атома серы. В процессе самосогласования было выполнено усреднение матрицы плотности по электронным состояниям, приближенно (в рамках ионной модели) соответствующим электронной конфигурации Ba2+S2– (Ba 6s0, S 3p6) в случае основного состояния X1Σ+ и конфигурации Ba+S– (Ba 6s1, S 3p5) для остальных рассмотренных состояний, a3Π, b3Σ+, A'1Π и A1Σ+.
При построении возбужденных конфигураций методами CCSD(T) и MRCI были учтены возбуждения электронов с МО, соответствующих валентным орбиталям 6s атома Ba и 3s, 3p атома S, а также внешним областям атомных остовов бария (5s, 5p) и серы (2s, 2p).
В большей части расчетов для атома бария был использован релятивистский псевдопотенциал (PP), замещающий 46 электронов атомного остова (1s–4d) [13], в сочетании с базисными наборами трех-, четырех- и пятиэкспонентного качества, cc-pwCVnZ-PP (n = T, Q, 5) [14]. При этом для атома S были использованы базисы aug-cc-pwCVnZ (n = T, Q, 5) [15, 16]. Далее эти базисы обозначены сокращениями wTZ, wQZ и w5Z.
Величины полной энергии молекулы, получаемые методом CCSD(T) с разными базисами, были экстраполированы к пределу полного базисного набора (CBS) по формуле [17]:
Здесь En – энергия, вычисленная в базисе с главным (кардинальным) числом n (например, у базиса wQZ число n = 4, а у базиса w5Z число n = 5); ECBS – энергия в пределе полного базисного набора. Коэффициент B легко определяется из соотношения: B = (En + 1 – En)/[(n + 1.5)–4 – (n + 0.5)–4].При вычислении энергии атомизации молекулы BaS вклад в энергию атома серы от спин-орбитального (SO) взаимодействия ΔESO был найден с использованием экспериментальных данных о тонкой структуре спектра атома серы [18] по формуле [19]:
(2)
$\Delta {{E}_{{{\text{SO}}}}} = --{{\Sigma }_{J}}(2J + 1){{E}_{J}}{\text{/}}{{\Sigma }_{J}}(2J + 1).$Ранее нами было показано [20], что для достаточно точного предсказания свойств соединений лантана (“соседа” бария по таблице Д.И. Менделеева) необходимо учитывать корреляцию 4d-электронов внешней части остова атома лантана. Поскольку в наших расчетах молекулы BaS для атома бария применялся псевдопотенциал, включающий в себя 4d-электроны, эффекты корреляции этих электронов мы находили с использованием для атома Ва полноэлектронного базисного набора Sapporo-DK3-QZP-2012 [21]. Этот базис мы дополнили примитивными гауссовыми функциями 3f2g1h с показателями экспонент αf = 8.9785, 3.5914, 1.4366; αg = 5.8562, 2.3425; αh = 4.1455, полученными в результате минимизации энергии основного состояния 1S0 атома Ba методом CISD с включением в корреляционную процедуру наряду с валентными и остовных 4d-электронов атома. В полноэлектронных расчетах молекулы BaS атом серы был описан базисом aug-cc-pVQZ-DK [15, 22]. Полученный таким образом набор далее обозначен как QZS. Скалярные релятивистские эффекты были учтены с применением гамильтониана Дугласа–Кролла–Гесса [23–26] третьего порядка (DK3).
Влияние спин-орбитального взаимодействия второго порядка (ΔSO) на свойства основного состояния молекулы BaS было учтено в полноэлектронных расчетах по программе DIRAC [27] методом CCSD(T) с четырехкомпонентным (4c) релятивистским гамильтонианом Дирака–Кулона [28, 29], 4c-DC-CCSD(T), в базисных наборах несгруппированных гауссовых функций Дайалла 31s24p16d8f2g для Ba и 19s12p7d4f для S [27, 30], далее обозначаемых как TZD. В корреляционную CCSD(T)-процедуру наряду с валентными электронами были включены остовные 5s- и 5p-электроны атома Ba. В пространство активных виртуальных орбиталей молекулы BaS в этих расчетах были включены все орбитали с энергией ниже +12 а.е. Чтобы выяснить роль электронной корреляции при вычислении спин-орбитальных эффектов ΔSO в молекуле BaS, результаты полноэлектронных расчетов методом 4c-DC-CCSD(T) были сопоставлены с результатами вычислений методом Хартри–Фока с гамильтонианом Дирака–Кулона, 4с-DC-HF. В расчетах методом 4с-DC-HF были использованы несгруппированные базисные наборы 35s30p19d7f5g1h для Ba и 25s15p8d6f4g для S [15, 18].
Спин-орбитальные эффекты были найдены также в расчетах с двухкомпонентным (2c) гамильтонианом [31] с использованием релятивистского псевдопотенциала остова для атома Ba [13] и 10-электронного PP остова атома серы [32]. В этих расчетах атом бария был описан расконтрактированным базисным набором функций 14s11p6d5f1g [14], а атом серы – несгруппированным набором примитивных функций, входящих в состав базиса aug-cc-pVQZ [15], из которого были исключены девять примитивных функций s-типа и четыре функции p-типа, обладающих самыми большими показателями экспонент (“наиболее тесно прижатых к ядру”). Исключенные функции описывают электронные оболочки атомного остова, замененные в этих расчетах псевдопотенциалом.
Эффектами высших порядков ΔHO мы называем корреляционные вклады, не учитываемые в приближении CCSD(T). Для основного электронного состояния молекулы BaS эти эффекты были вычислены вплоть до уровня CCSDTQ: ΔHO = CCSDTQ – CCSD(T). Величину ΔHO можно разбить на вклады от итерационного учета тройных возбуждений CCSDT – CCSD(T) и от учета четырехкратных возбуждений CCSDTQ – CCSDT. Далее для этих вкладов мы применяем обозначения ΔHOT–(T) и ΔHOQ–T соответственно. Вычисления эффектов ΔHO мы выполнили с использованием программы MRCC [33] в приближении замороженного остова. Вклады ΔHOT–(T) получали с применением трехэкспонентных базисов, а вклады ΔHOQ–T – с применением двухэкспонентных базисов (cc-pVnZ-PP (n = D, T) на атоме Ba [14] и aug-cc-pVnZ (n = D, T) на атоме S [15], далее обозначены как vDZ и vTZ соответственно).
Электронные состояния молекулы сульфида бария были также изучены методом MRCI в базисах wTZ и wQZ. Результаты были экстраполированы к пределу CBS по формуле (1) с n = 3 и 4. Наряду с триплетными состояниями a3Π и b3Σ+ этим методом были исследованы синглетные возбужденные состояния A'1Π и A1Σ+.
В MRCI-расчетах SO-взаимодействие было учтено путем построения матричного представления спин-орбитального оператора HSO, входящего в состав PP(Ba), в базисе чистых ΛS состояний (собственных векторов электростатического гамильтониана Hes), полученных методом MRCI, c последующей диагонализацией матрицы Hes + + HSO, представленной в этом базисе.
Расчеты спектроскопических параметров re (равновесное межъядерное расстояние, Å), ωe (частота колебания), ωexe (коэффициент ангармоничности), αe (постоянная колебательно-вращательного взаимодействия), а также констант De и βe (все в см–1), были выполнены методом Данхэма [34] по коэффициентам разложений потенциальной функции молекулы V(Δr) в ряд по степеням Δr = r – re с использованием значений V(Δr) в 11–13 точках ri, близких к re. С использованием полученных при такой обработке минимальных энергий рассчитаны адиабатические энергии возбуждения Te (см–1) и полная энергия атомизации TAE (ккал/моль).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Важнейшие вклады в свойства основного состояния молекулы сульфида бария показаны в табл. 1. Эффекты остовно-валентной корреляции, ΔCV, в этой таблице разделены на компоненты, обусловленные: а) корреляцией внешнеостовных 5s- и 5p-электронов атома бария, ΔCV(5s5p Ba); б) включением в число коррелируемых электронов наряду с валентными и остовными 5s- и 5p-электронами атома бария еще и 2s‑ и 2p-электронов атома серы, ΔCV(2s2p S); в) дальнейшим увеличением количества коррелируемых электронов за счет включения в корреляционную процедуру еще и 4d-электронов атома бария, ΔCV (4d Ba). Результаты численных экспериментов с последовательным расширением базиса свидетельствуют о достаточно быстрой и, как правило, плавной сходимости величин ΔCV(5s5p Ba) и ΔCV(2s2p S) к пределу полного базиса: wTZ → → wQZ → w5Z → CBS. Видно, что учет корреляции 5s- и 5p-электронов бария приводит к очень сильному изменению величин re, ωe и TAE: в CBS-пределе на 0.14 Å, 53 см–1 и 11 ккал/моль соответственно. Эффект корреляции остовных электронов атома серы заметно меньше, однако им не стоит пренебрегать в расчетах, претендующих на высокую точность. Учет корреляции 4d-электронов атомного остова Ba приводит к очень малым изменениям в спектроскопических постоянных, но заметно влияет на величину энергии атомизации молекулы (понижает ее более чем на 1 ккал/моль).
Таблица 1.
Вычисленные методом CCSD(T) вклады в параметры re, ωe и TAE молекулы BaS, X1Σ+, обусловленные учетом корреляции электронов атомных остовов ΔCV, спин-орбитального взаимодействия второго порядка ΔSO, корреляционных эффектов высших порядков ΔHO и нулевой колебательной энергии ΔZPE
| Параметр | Базис | Δre, Å | Δωe, см–1 | ΔTAE, ккал/моль |
|---|---|---|---|---|
| ΔCV(5s5p Ba) | wTZ | –0.0982 | +38.8 | +2.5 |
| wQZ | –0.1217 | +47.9 | +6.8 | |
| w5Z | –0.1320 | +51.0 | +9.2 | |
| CBS(Q5) | –0.1405 | +53.5 | +11.2 | |
| ΔCV(2s2p S) | wTZ | –0.0047 | +0.6 | +2.3 |
| wQZ | –0.0050 | +0.8 | +1.6 | |
| w5Z | –0.0040 | +0.6 | +0.8 | |
| CBS(Q5) | –0.0032 | +0.5 | +0.2 | |
| ΔCV (4d Ba) | QZS | –0.0002 | –0.2 | –1.1 |
| ΔSO | TZD | –0.0004 | +0.2 | +0.0 |
| ΔHOT–(T) | vTZ | –0.0003 | +0.3 | –0.5 |
| ΔHOQ–T | vDZ | +0.0036 | –2.8 | +0.5 |
| ΔZPE | CBS(Q5) | –0.5 |
Влияние спин-орбитального взаимодействия второго порядка на свойства молекулы BaS крайне мало. В табл. 1 представлены поправки ΔSO, рассчитанные методом 4c-DC-CCSD(T). Величины ΔSO, вычисленные в том же базисе и с тем же гамильтонианом, но без учета электронной корреляции (т.е. методом 4c-DC-HF), практически совпадают с результатами 4c-DC-CCSD(T)-расчетов. Хартри-фоковские расчеты величин re и ωe с применением PP бария дают поправки ΔSO, отличающиеся от результатов полноэлектронных расчетов лишь на 0.0002 Å и 0.1 см–1 соответственно.
Рассмотрим корреляционные эффекты высших порядков ΔHO в молекуле BaS. В первую очередь отметим весьма заметную величину вкладов ΔHOQ–T, обусловленных четырехкратными возбуждениями электронов. Кроме того, обращают на себя внимание очень малые величины поправок ΔHOT–(T). Этот результат свидетельствует о высокой точности корреляционных вкладов от тройных возбуждений, вычисляемых по теории возмущений.
Кривые потенциальной энергии молекулы BaS в основном и четырех низших возбужденных электронных состояниях показаны на рис. 1. Полученные методом MRCI/wTZ спектроскопические постоянные основного состояния молекулы, re = 2.5049 Å и ωe = 380.9 см–1, хорошо согласуются с результатами расчетов методом связанных кластеров и с экспериментальными данными (табл. 2). Такое согласие объясняется взаимной компенсацией ошибок в MRCI/wTZ величинах re и ωe, вызванных неполнотой wTZ-базиса и недостаточно полным учетом электронной корреляции в приближении MRCI. В то же время метод MRCI существенно занижает энергии электронного возбуждения молекулы BaS: Te = 6000–9000 см–1 (MRCI/wTZ), 7000–10 000 см–1 (MRCI/CBS), 12 000–15 000 см–1 (CCSD(T)/CBS). Отметим, что расширение базиса мало меняет разности вычисляемых методом MRCI энергий возбуждения в электронные состояния с одинаковым квантовым числом Λ, но с разной мультиплетностью: Te(A'1Π) – Te(a3Π) и Te(A1Σ+) – Te(b3Σ+). При переходе от базиса wTZ до CBS разность Te(A1Σ+) – Te(b3Σ+) изменяется на 312 см–1, а разность Te(A'1Π) – Te(a3Π) – всего лишь на 7 см–1.
Рис. 1.
Потенциальные кривые основного и низших возбужденных состояний молекулы BaS, рассчитанные в приближении MRCI/wTZ.
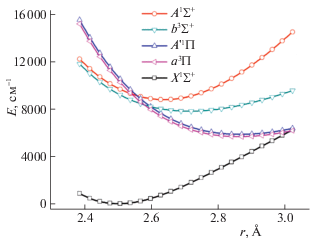
Таблица 2.
Теоретические и экспериментальные параметры re, ωe и Te молекулы BaS
| X1Σ+ | a3Π | A'1Π | b3Σ+ | A1Σ+ | ||
|---|---|---|---|---|---|---|
| re, Å | MRCI/CBS | 2.469 | 2.831 | 2.833 | 2.678 | 2.623 |
| CCSD(T)/CBS | 2.5079а | 2.8199а | 2.822б | 2.6593а | 2.605б | |
| Эксп. | 2.5073в | 2.813г 2.8159д |
2.813г 2.8217д |
2.635е 2.633г |
||
| ωe, см–1 | MRCI/CBS | 405 | 261 | 261 | 268 | 350 |
| CCSD(T)/CBS | 378.5а | 259.2а | 259б | 268.4а | 351б | |
| Эксп. | 379.4в | 259.6(5)г | 258.9(5)г 261.0(4)д |
294е 287.7(1)г |
||
| Te, см–1 | MRCI/CBS | 0 | 7007 | 7200 | 9092 | 9765 |
| CCSD(T)/CBS | 0 | 12 026а | 12 219б | 13 874а | 14 547б | |
| Эксп. | 0 | 11 744(10)ж 11 835(6)г |
12 186(10)ж 12 095(6)г 12 175.2д |
14 450е 14 498.6(1)г |
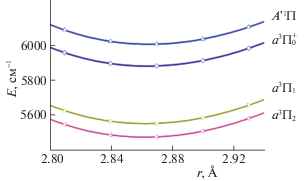
Рассмотрим предсказываемое теорией влияние спин-орбитального взаимодействия на характеристики возбужденных электронных состояний молекулы BaS. В первую очередь следует отметить асимметричный характер спин-орбитального расщепления энергетического уровня a3Π. Напомним, что при “обычном” спин-орбитальном расщеплении триплетных электронных состояний линейных молекул с волновой функцией симметрии Π (Λ = 1) спиновая компонента ΠΩ с Ω = 1 на энергетической шкале должна быть расположена посередине между двумя другими ΠΩ-компонентами с Ω = 0 и 2. Совсем иная ситуация имеет место в молекуле BaS. В изученном нами интервале межъядерных расстояний Ba–S (2.4–3.0 Å) компонента a3Π1 отстоит от компоненты a3$\Pi _{0}^{ + }$ приблизительно в 3 раза дальше, чем от компоненты a3Π2: E(a3$\Pi _{0}^{ + }$) – E(a3Π1) ≅ ≅ 330 см–1, E(a3Π1) – E(a3Π2) ≅ 90 см–1 (см. рис. 2). Такая асимметрия тонкой структуры энергетического уровня a3Π является следствием сильного спинового смешивания триплетного состояния a3Π с близлежащим синглетным состоянием A'1Π. Наши расчеты показали, что волновая функция спин-связанного состояния молекулы BaS, обозначаемого нами символом 3Π1, в действительности представляет собой смесь состояний – триплетного a3Π и синглетного A'1Π в пропорции 75% на 25%. Соответственно, волновая функция спин-связанного состояния, обозначаемого нами символом A'1Π, лишь на 75% состоит из “спин-несвязанного” синглетного состояния A'1Π. Остальные 25% обязаны своим происхождением триплетному состоянию a3Π. Еще одним следствием спин-смешивания состояний a3Π и A'1Π является заметное (на ∼125 см–1) возрастание энергии возбуждения состояния A'1Π. Энергии возбуждения остальных изученных нами электронных состояний молекулы BaS (b3Σ+ и A1Σ+) в результате учета SO-взаимодействия изменяются менее чем на 10 см–1. Спин-орбитальное взаимодействие мало влияет и на форму кривых потенциальной энергии возбужденных электронных состояний. В заключение обсуждения спин-орбитальных эффектов в молекуле BaS отметим, что асимметричность спин-орбитального расщепления электронного состояния a3Π этой молекулы была замечена ранее в работе [37].
Рис. 2.
Асимметрия спин-орбитального расщепления компонент 3Π2, 3Π1 и 3Π$_{0}^{ + }$ состояния a3Π вследствие частичного смешивания уровней 3Π1 и A'1Π (приближение SO-MRCI/wTZ).
Наилучшие теоретические оценки спектроскопических параметров re, ωe и Te молекулы BaS мы получили составным методом связанных кластеров для основного электронного состояния и методом CCSD(T)/CBS с включением корреляции остовных 5s- и 5p-электронов бария и 2s- и 2p-электронов серы для низших возбужденных триплетных состояний. Эти оценки выделены жирным шрифтом в табл. 2. Там же для сравнения показаны результаты расчетов в приближении MRCI/CBS, а также имеющиеся в литературе экспериментальные данные. Параметры возбужденных синглетных состояний A'1Π и A1Σ+ молекулы BaS, приведенные в строках “CCSD(T)”, оценены по формуле:
(3)
$\begin{gathered} {{P}_{S}}({\text{CCSD}}({\text{T}})) = {{P}_{T}}({\text{CCSD}}({\text{T}})) + \\ + \;{{P}_{S}}({\text{MRCI}})--{{P}_{T}}({\text{MRCI}}), \\ \end{gathered} $Параметры основного состояния молекулы BaS, найденные составным методом, прекрасно согласуются с опытными данными: отклонения от опыта составляют 0.0006 Å и 0.9 см–1 для re и ωe. Теоретические энергии электронных возбуждений молекулы согласуются с экспериментом в пределах 1 ккал/моль (350 см–1). Для состояния b3Σ+ в литературе имеется лишь оценка энергии возбуждения 13 000 ± 1000 см–1 [35]. В пределах своей погрешности эта оценка совпадает с величиной, рассчитанной нами методом CCSD(T)/CBS.
Энтальпия диссоциации D0 = TAE – ZPE молекулы BaS, полученная нами составным ab initio методом, составила 101.7 ккал/моль. Этот результат согласуется с величиной D0(BaS) = = 100.4 ± 3.6 ккал/моль, рекомендованной в справочнике [35].
Равновесный дипольный момент молекулы BaS в основном электронном состоянии X1Σ+ был с высокой точностью измерен в работе [39]: μe = = 10.86(2) D. Интересно сравнить этот результат с предсказаниями теории. Величину μe молекулы BaS (X1Σ+) мы получили в приближении CCSD(T)/CBS при re(Ba–S) = 2.5079 Å из первой производной от полной энергии молекулы по напряженности внешнего электростатического поля, вычисленной методом конечных разностей с шагом по напряженности поля ±0.0005 а.е. Рассчитанная нами величина μe = 10.90 D прекрасно согласуется с экспериментом [39]. Численным дифференцированием μe по межъядерному расстоянию r(Ba–S) в приближении CCSD(T)/CBS при r = re мы нашли величину первой производной дипольного момента молекулы BaS (X1Σ+): dμ/dr = 7.18 D/Å.
Предсказываемые теорией спектроскопические постоянные ωexe, αe, De и βe молекулы BaS в основном синглетном и низших возбужденных триплетных состояниях собраны в табл. 3. Наблюдается прекрасное согласие теоретических значений констант с результатами спектроскопических экспериментов, имеющимися в литературе. Этот факт является еще одним подтверждением высокой точности результатов нашей работы.
Таким образом, впервые с использованием высокоточных методов ab initio вычислены спектроскопические параметры основного (X1Σ+) и низших возбужденных триплетных (a3Π, b3Σ+) и синглетных (A'1Π, A1Σ+) состояний молекулы сульфида бария. Продемонстрирована очень высокая точность результатов расчетов методом связанных кластеров с учетом эффектов базиса, остовно-валентной корреляции электронов, спин-орбитального взаимодействия и корреляционных вкладов от электронных возбуждений высших порядков. Погрешность теоретического предсказания равновесного межъядерного расстояния и частоты колебания молекулы BaS в основном электронном состоянии не превышает 0.001 Å и 1 см–1 соответственно; энергия атомизации и относительные энергии возбужденных электронных состояний молекулы согласуются с опытными данными в пределах 1 ккал/моль (350 см–1). В целом результаты работы демонстрируют возможность неэмпирического описания спектроскопии и термохимии соединений тяжелых щелочноземельных элементов, по своей точности не уступающего результатам прецизионных экспериментальных измерений.
Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации (проект № FZZW-2020-0007).
Список литературы
Kalpana G., Palanivel B., Rajagopalan M. // Phys. Rev. B. 1994. V. 50. № 17. P. 12318. https://doi.org/10.1103/PhysRevB.50.12318
Zollweg R.J. // Phys. Rev. 1958. V. 111. № 1. P. 113. https://doi.org/10.1103/PhysRev.111.113
Morbi Z., Bernath P.F. // J. Mol. Spectrosc. 1995. V. 171. № 1. P. 210. https://doi.org/10.1006/jmsp.1995.1113
Werner H.-J., Knowles P.J., Knizia G. et al. MOLPRO, a package of ab initio programs. Version 2015.1. http://www.molpro.net.
Hampel C., Peterson K.A., Werner H.-J. // Chem. Phys. Lett. 1992. V. 190. № 1–2. P. 1. https://doi.org/10.1016/0009-2614(92)86093-W
Knowles P.J., Hampel C., Werner H.-J. // J. Chem. Phys. 1993. V. 99. № 7. P. 5219; ibid. 2000. V. 112. № 6. P. 3106 (erratum). https://doi.org/10.1063/1.465990, 10.1063/1.480886
Watts J.D., Gauss J., Bartlett R.J. // J. Chem. Phys. 1993. V. 98. № 11. P. 8718. https://doi.org/10.1063/1.464480
Werner H.-J., Knowles P.J. // J. Chem. Phys. 1988. V. 89. № 9. P. 5803. https://doi.org/10.1063/1.455556
Knowles P.J., Werner H.-J. // Chem. Phys. Lett. 1988. V. 145. № 6. P. 514. https://doi.org/10.1016/0009-2614(88)87412-8
Langhoff S.R., Davidson E.R. // Int. J. Quantum Chem. 1974. V. 8. № 1. P. 61. https://doi.org/10.1002/qua.560080106
Werner H.-J., Knowles P.J. // J. Chem. Phys. 1985. V. 82. № 11. P. 5053. https://doi.org/10.1063/1.448627
Knowles P.J., Werner H.-J. // Chem. Phys. Lett. 1985. V. 115. № 3. P. 259. https://doi.org/10.1016/0009-2614(85)80025-7
Lim I.S., Stoll H., Schwerdtfeger P. // J. Chem. Phys. 2006. V. 124. № 3. 034107. https://doi.org/10.1063/1.2148945
Li H., Feng H., Sun W. et al. // Mol. Phys. 2013. V. 111. № 14–15. P. 2292. https://doi.org/10.1080/00268976.2013.802818
Woon D.E., Dunning T.H. // J. Chem. Phys. 1993. V. 98. № 2. P. 1358. https://doi.org/10.1063/1.464303
Peterson K.A., Dunning T.H. // J. Chem. Phys. 2002. V. 117. № 23. P. 10548. https://doi.org/10.1063/1.1520138
Martin J.M.L. // Chem. Phys. Lett. 1996. V. 259. № 5–6. P. 669. https://doi.org/10.1016/0009-2614(96)00898-6
Kramida A., Ralchenko Yu., Reader J. et al. NIST Atomic Spectra Database. http://www.nist.gov/pml/data/asd.cfm.
Peterson K.A., Feller D., Dixon D.A. // Theor. Chem. Acc. 2012. V. 131. № 1. 1079. https://doi.org/10.1007/s00214-011-1079-5
Solomonik V.G., Smirnov A.N. // J. Chem. Theory Comput. 2017. V. 13. № 11. P. 5240. https://doi.org/10.1021/acs.jctc.7b00408
Noro T., Sekiya M., Koga T. // Theor. Chem. Acc. 2013. V. 132. № 5. 1363. https://doi.org/10.1007/s00214-013-1363-7
De Jong W.A., Harrison R.J., Dixon D.A. // J. Chem. Phys. 2001. V. 114. № 1. P. 48. https://doi.org/10.1063/1.1329891
Douglas M., Kroll N.M. // Ann. Phys. 1974. V. 82. № 1. P. 89. https://doi.org/10.1016/0003-4916(74)90333-9
Hess B.A. // Phys. Rev. A. 1986. V. 33. № 6. P. 3742. https://doi.org/10.1103/PhysRevA.33.3742
Wolf A., Reiher M., Hess B.A. // J. Chem. Phys. 2002. V. 117. № 20. P. 9215. https://doi.org/10.1063/1.1515314
Reiher M., Wolf A. // J. Chem. Phys. 2004. V. 121. № 22. P. 10945. https://doi.org/10.1063/1.1818681
Saue T., Visscher L., Jensen H.J.Aa. et al. DIRAC, a relativistic ab initio electronic structure program. Release 18 (2018). http://www.diracprogram.org.
Dyall K.G. // J. Chem. Phys. 1994. V. 100. № 3. P. 2118. https://doi.org/10.1063/1.466508
Visscher L. // Theor. Chem. Acc. 1997. V. 98. № 2–3. P. 68. https://doi.org/10.1007/s002140050280
Dyall K.G. // J. Phys. Chem. A. 2009. V. 113. № 45. P. 12638. https://doi.org/10.1021/jp905057q
Park Y.C., Lim I.S., Lee Y.S. // Bull. Korean Chem. Soc. 2012. V. 33. № 3. P. 803. https://doi.org/10.5012/bkcs.2012.33.3.803
Pacios L.F., Christiansen P.A. // J. Chem. Phys. 1985. V. 82. № 6. P. 2664. https://doi.org/10.1063/1.448263
Kállay M., Nagy P.R., Rolik Z. et al. MRCC, a quantum chemical program suite. http://www.mrcc.hu.
Dunham J.L. // Phys. Rev. 1932. V. 41. № 6. P. 721. https://doi.org/10.1103/PhysRev.41.721
Термодинамические свойства индивидуальных веществ. Справочное издание. Т. III. Кн. 1 / Под ред. Л.В. Гурвича. М.: Наука, 1981. 472 с.
Barrow R.F., Burton W.G., Jones P.A. // Trans. Faraday Soc. 1971. V. 67. P. 902. https://doi.org/10.1039/TF9716700902
Cummins P.G., Field R.W., Renhorn I. // J. Mol. Spectrosc. 1981. V. 90. № 2. P. 327. https://doi.org/10.1016/0022-2852(81)90131-4
Li G., Wang J.-G., Bernath P.F. // J. Mol. Spectrosc. 2012. V. 271. № 1. P. 10. https://doi.org/10.1016/j.jms.2011.10.003
Melendres C.A., Hebert A.J., Street K. // J. Chem. Phys. 1969. V. 51. № 2. P. 855. https://doi.org/10.1063/1.1672091
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии