

Журнал физической химии, 2021, T. 95, № 3, стр. 443-451
Особенности температурной зависимости скорости окисления и распада горючих газовых веществ
В. В. Азатян a, *, С. К. Абрамов b, И. А. Болодьян c, Д. М. Гордиенко c, Н. П. Копылов c, В. М. Прокопенко b, Ю. Н. Шебеко c
a Российская академия наук, Институт химической физики им. Н.Н. Семенова
Москва, Россия
b Российская академия наук, Институт структурной макрокинетики и проблем материаловедения
им. А.Г. Мержанова
Московская область, Черноголовка, Россия
c ФГУ ВНИИПО МЧС России
Москва, Россия
* E-mail: vylenazatyan@yandex.ru
Поступила в редакцию 05.05.2020
После доработки 25.05.2020
Принята к публикации 27.05.2020
Аннотация
Показано, что процессы окисления газов вне области воспламенения являются изотермическими цепными процессами без прогрессирующего самоускорения. Определен функциональный характер температурной зависимости скорости реакций в этих условиях. При нагревании до критической температуры квазистационарный режим реакции сменяется цепной лавиной, в ходе развития которой становятся заметными саморазогрев и его роль. Получены аналитические выражения, описывающие кинетику процесса вне области воспламенения и в режиме горения. Показано различие температурной зависимости скорости в этих режимах. Выяснен закон температурной зависимости скорости неразветвленно-цепных процессов в режимах воспламенения и взрыва.
Зависимость скорости реакций от температуры определяет важнейшие их закономерности в неизотермических условиях. Осуществляя положительную обратную связь между температурой горючей смеси и скоростью процесса, температурная зависимость обеспечивает возгорание при нагревании, определяет ускорение реакций под воздействием выделяющегося тепла, переход горения во взрыв и в детонацию. Характер влияния температуры на кинетику реакций определяется, прежде всего, особенностями температурной зависимости скорости процесса, разными при разных механизмах процесса. До недавнего времени было общепринятым считать, что процессы горения газов при давлениях выше сотых долей атмосферного давления в условиях значительного саморазогрева протекают без заметной роли реакционных цепей [1–8]. В настоящее время, однако, известно, что газофазные процессы горения протекают при определяющем участии реакционных цепей не только при давлениях в десятки и сотни раз ниже атмосферного давления, в отсутствие заметного саморазогрева, но также при атмосферном и повышенных давлениях, в любом температурном режиме (например, [9–12]). Соответственно все особенности горения, в том числе, зависимость скорости реакции от температуры, определяются законами цепных реакций. В настоящей работе на примере модельного процесса окисления водорода рассматриваются особенности и роль температурной зависимости скорости реакций в различных кинетических режимах.
КОРЕННОЕ РАЗЛИЧИЕ ТЕМПЕРАТУРНЫХ ЗАВИСИМОСТЕЙ СКОРОСТИ РЕАКЦИИ И КОНСТАНТЫ СКОРОСТИ
В теории процессов газофазного горения было принято представлять неизотермический химический процесс одностадийной реакцией с учетом также уравнения теплового баланса, т.е. учитывалось только тепловое самоускорение. Важнейшие положения теории горения относящиеся к воспламенению, распространению пламени и детонации, основаны на этой модели и на представлениях, согласно которым, температурная зависимость скорости реакций соответствует закону Аррениуса (например, [1–8]). До последнего времени было принято также считать, что зависимость скорости реакции и ее константы от температуры, зависят тем сильнее, чем больше энергия активации (например, [2–4, 8]).
В настоящее время, однако, установлено, что эти представления неправильные и поэтому противоречат экспериментам. Было показано, что химические процессы, лежащие в основе горения, взрыва и детонации, это – цепные реакции с присущими им специфическими закономерностями (например, [10–13]). Было обращено также внимание на то, что функция Аррениуса относится не к скорости реакции, а к константе скорости. Неоправданное допущение о соответствии температурной зависимости скорости функции Аррениуса противоречит, прежде всего, фундаментальному закону действующих масс, согласно которому, скорость реакции равна произведению константы скорости и концентраций реагентов, т.е. произведению разных функций. Например, скорость реакции первого кинетического порядка равна:
где С(T, t) – концентрация, Т – температура, t – время, k(T, t) – константа корости.В ходе горения функции k(T, t) и С(T, t) изменяются по разным законам. Отождествление температурных зависимостей величин W(T, t) и k(T, t) означает неоправданное игнорирование сильного изменения множителя С(T, t). Кроме того, экспоненциальная функция Аррениуса не способна описывать и не описывает присущую процессам горения специфическую, очень сильную зависимость скорости от температуры [11–13]. По той же причине неправильно встречающееся нередко представление зависимости характеристического времени реакции от температуры экспоненциальной функцией [14, 15].
Отождествление функции Аррениуса с температурной зависимостью скорости реакции – одна из причин того, что наблюдаемые большие скорости реакций горения и их сильная температурная зависимость, особенно в режимах взрыва и детонации, длительное время не находили объяснения. Неприменимость аррениусовской функции к описанию температурной зависимости скорости иллюстрирует следующий пример. При нагревании стехиометрической смеси Н2 с О2 при 1 атм от 823 до 853 К константа скорости реакции между исходными молекулами, характеризующейся энергией активации 225 кДж/моль, увеличится лишь на $ \approx $5%. Рост скорости этой реакции с температурой определяется ростом константы скорости. В данном случае скорость возрастет даже немного меньше, поскольку при повышении температуры начальные концентрации исходных реагентов несколько снижаются. Между тем, эксперимент показывает, что при указанном выше нагреве смесь воспламеняется и выгорает за доли миллисекунды, т.е. реакция ускоряется во много тысяч раз больше, и, значит, скорость от температуры зависит несравненно сильнее, чем экспоненциальная функция Аррениуса.
Также неправильны представления о более сильной температурной зависимости W(T) и k(T) при большей энергии активации. Действительно, мерой температурной зависимости константы скорости служит абсолютная величина ее изменения при единичном изменении температуры: $\frac{{\partial k}}{{\partial T}}~$ [11, 13]. Из выражения для константы скорости:
следует, что(3)
$\beta = \frac{{\partial k}}{{\partial T}} = {{k}_{0}}{{e}^{{ - ~\frac{E}{{RT}}}}}\frac{E}{{R{{T}^{2}}}} = k\frac{E}{{R{{T}^{2}}}}.$Рис. 1.
Функции k/k0= e–E/RT при различных значениях энергии активации: 1 – 25, 2 – 150, 3 – 225 кДж/моль. На вставке показан участок высоких температур.
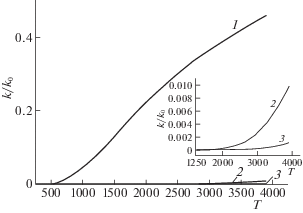
В процессах же горения, как было показано выше, скорость возрастает очень резко, и функция ${{e}^{{ - E/RT}}}$, независимо от величины энергии активации, не описывает наблюдаемого сильного самоускорения реакции.
Численное моделирование горения лишь формально описывает отдельные характеристики процесса в рамках принятой совокупности элементарных реакций, тепловых эффектов и констант скорости. При этом используемые реакционные схемы даже горения водорода с наиболее изученным механизмом не содержат все необходимые стадии, в том числе, реакции гетерогенного обрыва цепей. Между тем, реакции обрыва блокируют цепной путь, тормозят процесс, влияют на градиенты. Если при горении органических соединений образуется сажа, то гетерогенными являются реакции атомов и радикалов с диспергированными частицами. В некоторых же публикациях нереальны реакционные схемы, кинетические уравнения противоречат записанным реакциям. Например, в работе [17] моделирование горения метана проведено без учета реакций свободных атомов и радикалов, без которых газофазное горение, как известно, невозможно [9–13]. Записанным тримолекулярным реакциям 2Н2 с О2 и 2СО с О2 приписаны константы скорости реакций второго кинетического порядка с нарушением размерности. Это привело также к завышению рассчитанных скоростей реакции и тепловыделения во много тысяч раз.
ЦЕПНОЙ ХАРАКТЕР РЕАКЦИЙ ОКИСЛЕНИЯ ГАЗОВ ВНЕ ОБЛАСТИ ВОСПЛАМЕНЕНИЯ
Одна из основных закономерностей процессов горения, определяемых ростом скорости реакции при повышении температуры, – широко распространенные в технике и в быту возгорание и взрыв горючих газов при нагревании. Значимость проблемы выяснения механизма воспламенения и взрыва горючих газов при разогреве определяется не только проблемой взрывобезопасности, но в большей мере тем, что воспламенение, вызванное нагреванием тепловым потоком из прилегающей зоны горения, является необходимой стадией распространения пламени, представляющего собой послойное возгорание. Аналогично, воспламенение, вызванное нагреванием подходящей ударной волной, – необходимая стадия детонации. Возгорание топлива, вызванное повышением температуры под воздействием адиабатического сжатия, осуществляется, как известно, также в дизельных силовых установках.
Приводимое ранее в теории горения объяснение воспламенения газов при нагревании основывалось на модели одностадийной реакции валентно-насыщенных молекул [2–8, 18]. В работах [10–12], однако, было показано, что реакции валентно-насыщенных соединений между собой в силу очень больших энергий активации настолько медленны, что выделяющееся тепло практически полностью отводится теплопроводностью. Поэтому такие реакции не могут протекать и не протекают в режиме горения. В отсутствие реакций свободных атомов и радикалов сильный нагрев горючего газа может быть осуществлен только внешним источником тепла (например, ударной волной или прилегающей зоной горения в распространяющемся пламени). Однако, как было отмечено выше, межмолекулярным реакциям присущи не только крайне малые скорости, но и очень слабая зависимость их скорости от температуры [10, 11]: абсолютный рост скорости при нагревании даже до 4000 К незначителен. Поэтому теория, учитывающая реакции только валентно-насыщенных соединений, не способна объяснить и не объясняет наблюдаемые воспламенение, взрыв, вызванные нагреванием, не объясняет также распространение пламени и детонацию газов. Она только исходит из факта протекания этих процессов.
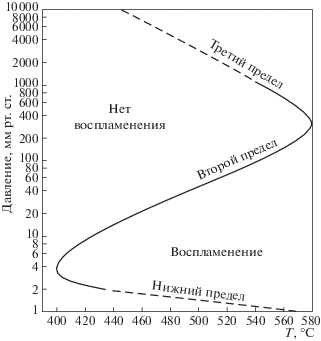
В настоящей работе кинетика воспламенения газа, вызванного нагреванием, рассматривается на примере модельного цепного процесса окисления водорода. На рис. 2 приведены границы области самовоспламенения стехиометрической смеси Н2 с О2 в термостатированном при каждой заданной температуре замкнутом сферическом кварцевом реакторе, сполоснутом в различных опытах водными растворами KCl или смеси KCl и K2B4O7 [19]. Данные рис. 3 из работы [19] соответствуют цепной природе очень медленного процесса вне области воспламенения. В указанном реакторе в смеси 2Н2 + О2 при 485 Торр и 570°С (843 К), т.е. на 2 К ниже критической температуры (рис. 2), давление очень медленно падает вследствие уменьшения числа молей смеси в суммарной реакции
Саморазогрев настолько слабый, что соответствующий ему рост давления не компенсирует даже малое падение давления. Для выяснения характера кинетического закона процесса определим сначала его скорость и сравним со скоростью элементарной реакции Н2 с О2. По стехиометрии реакции (A), падение давления смеси ΔP равно снижению парциального давления кислорода: $~\Delta {{p}_{{{{{\text{O}}}_{2}}}}}$. Из рис. 3 видно, что при 485 Торр и 570°С (843 К) скорость падения давления в момент времени 0.4 мин равна –30 Торр/мин, т.е. –0.50 Торр/с. В пересчете на концентрацию скорость реакции равна: $W_{{{\text{ст}}}}^{{{\text{эксп}}}}$ = –9.6 × 10–9 моль/(см3 с). Это в сотни раз больше, чем скорость элементарной реакции: Действительно, скорость реакции (0) равна(4)
${{\omega }_{0}} = - {{\left\{ {\frac{{d[{{{\text{O}}}_{2}}]}}{{dt}}} \right\}}_{{\text{0}}}} = ~{{k}_{0}}[{{{\text{H}}}_{2}}][{{{\text{O}}}_{{2~}}}] = 2{{k}_{0}}{{[{{{\text{O}}}_{2}}]}^{2}},$Рис. 2.
Три предельных давления самовоспламенения стехиометрической смеси водорода с кислородом по данным работы [8].
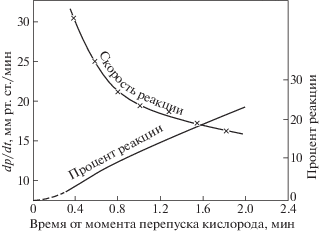
Константе скорости реакции (0), равной 6 × × 1013$\left( { - \frac{{26\,500K}}{T}} \right)$ см3 моль–1 с–1 [20, 21], и концентрации О2, равной в данных условиях 3.1 × × 10–6 моль/см3, соответствует скорость реакции (0) при 843 К, близкая к –2 × 10–11 см3 моль–1 с–1. Это в 480 раз меньше приведенной выше измеренной величины $W_{{{\text{ст}}}}^{{{\text{эксп}}}}$. Такое сильное превышение скорости брутто-процесса над скоростью элементарной реакции (0) означает, что вне области воспламенения процесс протекает не по межмолекулярному пути, а при активном участии свободных атомов и радикалов – по цепному пути.
Для выяснения роли саморазогрева в этом цепном процессе сравним характеристические времена реакции (${{t}_{p}}$) и теплоотвода (tT). Время расходования исходных реагентов рассчитаем по известной формуле реакции второго порядка в стехиометрической смеси, получающейся интегрированием уравнения (4), где, однако, вместо константы скорости ${{k}_{0}}$ элементарной реакции (0) входит эффективная константа скорости брутто-процесса ${{k}^{{{\text{эфф}}}}}$:
(5)
$t = \frac{1}{{2{{k}_{{{\text{эфф}}}}}}}\left( {\frac{1}{C}~ - - ~\frac{1}{{{{C}_{0}}}}} \right).$Величину ${{k}^{{{\text{эфф}}}}}$ определим по выражению скорости брутто-процесса:
(6)
${{W}_{{{\text{эксп}}}}} = - \frac{{d[{{{\text{O}}}_{2}}]}}{{dt}} = ~{{k}_{{{\text{эфф}}}}}[{{{\text{H}}}_{2}}][{{{\text{O}}}_{{2~}}}].$Время теплоотвода рассчитывается по уравнению Эйнштейна–Смолуховского [22] с учетом сферической формы реактора:
где χ – коэффициент температуропроводности, который на ≈20% больше коэффициента диффузии D [23], r – радиус реактора, равный 3.7 см.При 293 К и 100 кПа коэффициент диффузии О2 в смеси с Н2 равен 0.69 см2/с [24] и, значит, ${{\chi }_{0}} \approx 0.84$ см2/с. Учитывая зависимость χ от давления и температуры:
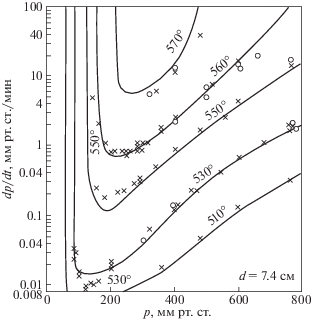
получаем величину $\chi $, равную 7.14 см2/с при 843 К и 485 Торр. Согласно выражению (7), этим величинам $\chi $ и r соответствует значение ${{t}_{T}}$ ~ 0.64 с. Таким образом, характеристическое время реакции в ~800 раз больше, чем время теплоотвода, и значит, подпредельная реакция протекает практически без саморазогрева, и сильное превышение скорости изотермического расходования О2 над скоростью элементарной реакции (0) вызвано именно цепным характером процесса. По мере приближения начальных величин давления и температуры к критическому условию воспламенения скорость подпредельной изотермической реакции, прогрессивно возрастает (рис. 4), все сильнее превышая скорость реакции (0). Абсолютная величина роста скорости межмолекулярной реакции незначительна, поскольку крайне мала константа ее скорости, как это видно также из рис. 1.Рис. 4.
Начальные скорости реакции вне области самовоспламенения при различных начальных температурах и давлениях.
Коренное отличие кинетики реального процесса вне области воспламенения, от принимаемой традиционно гипотетической межмолекулярной реакции проявляется также в аномально большой “энергии активации” процесса, получаемой при допущении о соответствии температурной зависимости экспериментальной величины скорости закону Аррениуса и рассчитываемой по уравнению:
где W1 и W2 – скорости при температурах Т1 и Т2.По данным рис. 4 (рис. 2.7 монографии [19]) начальные скорости падения давления при 843 и 833 К равны соответственно – 50 и – 4 Торр/мин. При подстановке этих величин в выражение (5), получается эффективная энергия активации, равная 353 ккал/моль. Это на сотни ккал/моль больше реальных энергий активации реакций между валентно-насыщенными соединениями. Более того, определенная таким способом “энергия активации” зависит от начальных условий. Такая “энергия активации”, и ее зависимость от давления служат дополнительными показателями несоответствия реальной температурной зависимости скорости реакции закону Аррениуса.
СПЕЦИФИКА ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ СКОРОСТИ РЕАКЦИИ, ОБЕСПЕЧИВАЮЩАЯ ВОСПЛАМЕНЕНИЕ ПРИ НАГРЕВАНИИ
Характер температурной зависимости скорости, приводящей к воспламенению при нагревании, а также цепная лавина внутри области воспламенения, определяются механизмом процесса со следующими основными стадиями [1, 11]:
(VI)
${\text{H}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{2}} = {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}} + {\text{H}},$Ниже рассматривается соответствующее приведенной реакционной схеме уравнение, описывающее зависимость скорости подпредельной реакции от температуры, а также переход процесса в режим прогрессивного цепного самоускорения. Из этой схемы видно, что скорость расходования О2, т.е. скорость цепного процесса равна:
(6а)
$\begin{gathered} W = - d[{{{\text{O}}}_{2}}]{\text{/}}dt = {{\omega }_{0}} + \{ {{k}_{1}} + ~{{k}_{5}}[{\text{M}}]\} [{{{\text{O}}}_{2}}][{\text{H}}] = \\ = {{\omega }_{0}} + ~{{k}_{c}}\left[ {{{{\text{O}}}_{2}}} \right]\left[ {\text{H}} \right], \\ \end{gathered} $На основе приведенного выше механизма с использованием метода частичных стационарных концентраций [1, 18] получается следующее выражение для скорости изменения концентрации атомов Н, осуществляющих разветвление цепей по реакции (I):
(10)
$\frac{{d[{\text{H}}]}}{{dt}} = {{\omega }_{0}} + (f - g)[{\text{H}}] = {{\omega }_{0}} + {\varphi }[{\text{H}}].$(11)
$f = 2{{k}_{1}}[{{{\text{O}}}_{2}}] = 2k_{1}^{0}{{e}^{{ - ~\frac{{{{E}_{1}}}}{{RT}}}}}[{{{\text{O}}}_{2}}],~$(14)
$W = - \frac{{d[{{{\text{O}}}_{2}}]}}{{dt}} = {{\omega }_{0}}\left\{ {1 + \frac{{{{k}_{1}}[{{{\text{O}}}_{2}}]}}{{f - g}}[{{е}^{{(f - g)t}}} - 1]} \right\}.$(15)
${{W}_{{{\text{cт}}}}} = - \frac{{d[{{{\text{O}}}_{2}}]}}{{dt}} = {{\omega }_{0}}\left\{ {1 + \frac{{{{k}_{1}}[{{{\text{O}}}_{2}}]}}{{g - f}}[1 - {{{\text{е}}}^{{ - (g - f)t}}}]} \right\}.$Согласно выражению (15), при временах t, превышающих 4/(g – f), второй член квадратной скобки намного меньше единицы и, величина W становится не зависящей от времени. Из приведенной выше экспериментальной величины скорости реакции следует, что за одну секунду расходуется лишь ~0.3% О2, т.е. за интересующий нас промежуток времени 0.015 с, исходные реагенты расходуются не более, чем на 0.005%. Поэтому их концентрации с высокой точностью можно считать зависящими только от g и f при каждой данной температуре.
Рост ${{\omega }_{0}}$, вызванный нагреванием, крайне мал, поскольку мала сама величина ${{\omega }_{0}}$. Величины же ${{k}_{1}}$ и, значит, f, благодаря своим большим значениям, возрастают сильнее. При этом, поскольку скорость обрыва и, значит, g от температуры зависят слабо, то при повышении температуры возрастающая величина f приближается к g, знаменатель дроби уменьшается, и стационарная при каждой данной температуре величина W гиперболически возрастает. При дальнейшем нагревании и приближении величины f к g$~$второй член квадратной скобки приближается к единице, проявляется стремление к нулю квадратной скобки так же, как и знаменателя. В итоге отношение скобки к знаменателю стремится к величине t, согласно правилу Лопиталя, и с учетом выражения (7) получается:
(16)
${{W}_{{{\text{ст}}}}} = {{\omega }_{0}}\left[ {1 + \frac{1}{{2\left( {\frac{g}{f}~ - 1} \right)}}} \right].$ЦЕПНОЙ ХАРАКТЕР ПЕРЕХОДА КВАЗИСТАЦИОНАРНОЙ РЕАКЦИИ В РЕЖИМ ВОСПЛАМЕНЕНИЯ
Из рис. 2 видно, что при давлении 485 Торр температура на третьем пределе воспламенения находится между 573 и 575°С. Непосредственно под третьим пределом в результате даже небольшого нагревания (на 3–5 К) и соответствующего роста величины f разность f – g становится положительной. Вследствие этого характер зависимости W от времени претерпевает качественное изменение: скорость во времени возрастает в соответствии с уравнением (15), гиперболическая зависимость скорости реакции от (g – f) сменяется экспоненциальной зависимостью от (f – g)t, начинается цепная лавина. Изменяется также характер температурной зависимости скорости: при f > g функция температурной зависимости содержит фактор Больцмана в положительной экспоненте [11, 25, 26] (см. ниже).
При изучении роли саморазогрева и раздельно роли цепной лавины в возникновении воспламенения следует рассматривать самые начальные времена реакции, за которые успевает реагировать лишь 0.3–0.5% исходных веществ. Как это показывает также численное решение системы кинетических уравнений совместно с уравнением теплового баланса, при таких временах саморазогрев крайне мал: не превышает несколько градусов [27], т.е. меньше 0.5%, поскольку очень малы как количество выделившегося тепла, так и скорость тепловыделения. Без цепной лавины такой саморазогрев не может привести к воспламенению, воспламенение является цепным. Цепно-тепловым же является горение, в котором наряду с цепной лавиной реализуется также условие теплового воспламенения: от повышения температуры тепловыделение ускоряется больше, чем теплоотвод.
Показателем того, что воспламенение при атмосферном давлении начинается цепной лавиной, служат экспериментальные и расчетные данные, (например, [11, 28]), показывающие, что после инициирования воспламенения хемилюминесценция регистрируется раньше, чем рост давления. Кроме того, максимальная скорость роста хемилюминесценции достигается раньше максимальной скорости роста давления. Серией экспериментальных (например, [9–11, 28–30]) и расчетных [11] исследований показано, что переход горения во взрыв и в детонацию также определяется цепным механизмом реакций. Таким образом, в отличие от модели реакции только между исходными молекулами, использованной в теории теплового горения и не способной объяснить воспламенение при нагревании, выявление и учет цепной природы реакции вне области воспламенения позволяют объяснить возгорание газа, вызванное внешним источником тепла. Благодаря этому, находят объяснение начальные стадии воспламенения в каждом очередном слое горючего газа в распространяющемся пламени и в детонации. Воспламенению способствует поступление атомов и радикалов в свежую смесь из зоны горения.
ЗАКОН ЭКСПОНЕНТЫ В ПОЛОЖИТЕЛЬНОЙ ЭКСПОНЕНТЕ
Резкое самоускорение реакций горения и взрыва указывает на то, что температурная зависимость скорости реакции коренным образом отличается от традиционно используемой экспоненциальной функции в формуле Аррениуса. Специфический характер этой зависимости определяется тем, что в каждый момент времени и при каждых данных величинах f и g скорость изменения концентрации НЦ находится в положительной обратной связи с самой концентрацией этих активных частиц, как это видно, например, из уравнения (10). Это значит, что величина [Н] возрастает во времени экспоненциально даже при постоянной температуре. В эту экспоненту в качестве множителя времени t входит разность f ‒ g. Величина f содержит константу скорости разветвления k1 с ее фактором Больцмана в соответствии с выражением (11). Таким образом, при f > g величины [Н] и W зависят от температуры по закону экспоненты с положительным показателем степени [11]. Поскольку функциональная зависимость “экспоненты в положительной экспоненте” осуществляется при каждой данной температуре, то она выполняется в ходе горения.
Резкое ускорение процесса при повышении температуры вызвано сильным ускорением реакций (1) и (2) благодаря большим величинам констант их скорости. В этих реакциях происходит экспоненциально ускоряющееся размножение активных частиц и, значит, сильно ускоряющееся расходование исходных реагентов, т.е. усиливающееся ускорение горения. При временах t > t0 ≅ ≅ 2.5/φ, когда в уравнениях (10) и (11) уже можно пренебречь величиной ${{\omega }_{0}}$, интегрирование уравнения (6) с учетом выражения формулы (7) и температурной зависимости k1 приводит к следующей зависимости $\left[ {\text{H}} \right]~$ от температуры и времени:
(17)
$[{\text{H}}] = {\text{\;}}{{[{\text{H}}]}_{0}}\exp \mathop \smallint \limits_{{{t}_{0}}}^t \left[ {{{f}_{0}}\exp \left( { - \frac{{{{E}_{{{\text{pазв}}}}}}}{{RT}}} \right) - g} \right]dt.$При подстановке [H] из выражения (18) в уравнение скорости (6а) получаем:
(18)
$W{\text{/}}[{{{\text{O}}}_{2}}] = {{k}_{1}}{{[{\text{H}}]}_{{0{\text{\;}}}}}\exp \mathop \smallint \limits_{t{\text{о}}}^t [{{f}_{0}}\exp ( - E{\text{/}}RT) - g]{\kern 1pt} dt.$Описанные закономерности цепного горения относятся также к другим горючим газам, в том числе к метану, синтез-газу, пропану, гидразину.
После выявления цепной природы процессов горения, взрыва и детонации газов [9–13, 30] стало очевидным, что реальное понимание и обоснованная интерпретация тех или иных закономерностей любых режимов горения возможны только при выяснении механизма наблюдаемых закономерностей с учетом законов неизотермических цепных процессов. Этими законами определяется описанная выше специфика температурной зависимости скорости реакций вне области воспламенения и в режиме горения. Численное решение уравнений, лишь констатирующее согласие решений с той или иной наблюдаемой закономерностью, такой задаче не соответствует даже при правильных уравнениях и входных параметрах.
Таким образом, в хорошем согласии с [9–13, 27], воспламенение, распространение пламени и детонация протекают при последующей реализации перечисленных ниже процессов. При нагревании под воздействием тепла внешнего источника в смеси, находящейся первоначально вне области воспламенения, предельная стационарная величина скорости цепной реакции ${{W}_{{{\text{ст}}}}}$ возрастает в соответствии с гиперболическими зависимостями (15), (16) от разности $g - f$ до достижения критического значения, при котором величина f становится больше g, и происходит переход процесса в режим цепного воспламенения. С этого момента осуществляется рост скорости с температурой по закону экспоненты в положительной экспоненте.
ЗАКОН ЭКСПОНЕНТЫ В ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ СКОРОСТИ НЕРАЗВЕТВЛЕННО-ЦЕПНЫХ ПРОЦЕССАХ
Неразветвленно-цепные процессы (например, реакция хлора с водородом, распад гидразина и ацетилена) часто протекают в режиме взрыва. Как было показано выше, рост величин даже большой константы скорости сам по себе не может привести к скоростям реакции, необходимым для взрыва. Приведенное ниже рассмотрение показывает, что такие скорости достигаются в результате того, что закон экспоненты с положительным показателем степени реализуется также в неразветвленно-цепных процессах, которым соответствует следующая реакционная схема:
где х и у – атомы и радикалы, А и В – исходные молекулярные реагенты, Р – конечный продукт, а также уравнения, относящиеся к скорости цепной реакции и концентрации y(19)
$W = - \frac{{d[{\text{А}}]}}{{dt}} = \frac{{{{\omega }_{0}}}}{2} + {{k}_{{\text{д}}}}[{\text{y\;}}][{\text{B}}],~$(20)
$\frac{{d[y]}}{{dt}} = {{\omega }_{0}} + {{k}_{{\text{г}}}}[{\text{x}}][{\text{A}}] - - {{k}_{{\text{д}}}}[{\text{y}}][{\text{B}}] - - {{k}_{{\text{ж}}}}[{\text{y}}].$В развивающемся процессе концентрации всех НЦ возрастают. В этих условиях справедливо соотношение: [x] = b[y], где b – положительная величина, отражающая симбатность роста концентраций частиц x и y. Подставив b[y], вместо [x] в уравнение (20) и вынеся [y] за скобки, получаем:
(21)
$\frac{{d[{\text{y}}]}}{{dt}} = {{\omega }_{0}} + \{ {{k}_{{\text{г}}}}b[{\text{A}}] - {{k}_{{{\text{\;д}}}}}[{\text{B}}] - {{k}_{{\text{ж}}}}]\} [{\text{y}}] = {\Psi }[{\text{y}}].$Работа выполнена при финансовой поддержке РФФИ (грант № 18-03-00489), а также Минобрнауки России по государственному контракту № 05.607.21.0304 (идентификатор контракта RFMEFI60719X0304) “Разработка эффективных способов химического ингибирования гомогенных и гетерогенных процессов горения и создание демонстрационного образца детонационного конвертора техногенных отходов” в рамках Федеральной целевой программы “Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России на 2014–2020 годы”, а также в рамках государственного задания ИСМАН (тема 45.1).
Список литературы
Семенов Н.Н. Избранные труды. М.: Наука, 2005. Т. 3. 499 с.
Зельдович Я.Б., Баренблат Г.И., Либрович В.Б., Махвиладзе Г.М. Математическая теория горения. М.: Наука, 1980. 478 с.
Кондратьев В.Н., Никитин Е.Е. Химические процессы в газах. М.: Наука, 1981. 262 с.
Химическая энциклопедия. Горение. М.: Сов. энциклопедия, 1988. Т. 1. С. 594.
Физическая энциклопедия. Горение. М.: Сов. энциклопедия, 1988. Т. 1. С. 515.
Мержанов А.Г., Хайкин Б.И. Теория волн горения в гомогенных средах. Черноголовка: Изд-во Ин-та структурной макрокинетики РАН, 1992. 160 с.
Большой энциклопедический словарь. Физика. Статья Взрыв. М., 1998. С. 73.
Франк-Каменецкий Д.А. Основы макрокинетики, диффузия, теплопередача в химической кинетике. Долгопрудный: Интеллект, 2008. 407 с.
Азатян В.В., Павлов В.А., Шаталов О.П. // Кинетика и катализ. 2006. Т. 40. № 6. С. 835.
Азатян В.В. // Журн. физ. химии. 2011. Т. 85. № 8. С. 1405.
Азатян В.В. Цепные реакции в процессах горения, взрыва и детонации газов. Черноголовка: Изд-во ИПХФ РАН, 2017. 448 с.
Азатян В.В. // Кинетика и катализ. 2015. Т. 56. № 1. С. 3.
Азатян В.В., Болодьян И.А., Копылов Н.П. и др. // Журн физ. химии. 2018. Т. 92. № 5. С. 701.
Нетлетон М. Детонация в газах. М.: Мир, 1989. 278 с.
Borghi R., Fruchard N. // Progr. in Astronautics and Aeronautics. 1997. V. 173. P. 221.
Азатян В.В., Павлов В.А., Шаталов О.П. // Кинетика и катализ. 2005. Т. 46. № 6. С. 835.
Frolov S.M., Shamshin I.V., Dubrovsrii F.V., Medvedev S.N. // Transient Combustion and Detonation. Moscow: TORUS PRESS, 2014. P. 204.
Семенов Н.Н. О некоторых проблемах химической кинетики и реакционной способности. М.: Изд-во АН СССР, 1958. 686 с.
Lewis B., von Elbe G. Combustion, Explosions and Flame in Gases. N.Y.–L.: Acad. Press, 1987.
Baulch D.L., Bowman C.T., Cobos C.J. et al. // J. Phys. Chem. Ref. Data. 2005. V. 34. № 3. P. 757.
Srinivasan N.K., Michael J.V., Harding L.B., Klipperstain S.J. // Combust. Flame. 2007. V. 149. № 1–2. P. 104.
Ландау Л.Д., Ахиезер А.И., Лифшиц Е.М. Курс общей физики. М.: Наука, 2014. 379 с.
Телеснин Р.В. Молекулярная физика. М.: Высшая школа, 1973. 360 с.
Чепмен С., Каулинг Т. Математическая теория неоднородных газов. Пер. с англ. М.: Изд-во иностр лит., 1960. 510 с. (Sydney Chapman, T. G. Cowling. The Mathematical Theory of Non-uniform Gases. 1970. Cambridge University Press, 1.423 p.)
Азатян В.В. // Кинетика и катализ. 1976. Т. 17. № 2. С. 533.
Азатян В.В ., Шавард А.А., Калканов В.А., Мержанов А.Г. // Хим. физика. 1987. Т. 6. № 12. С. 1696.
Азатян В.В., Андрианова З.С., Иванова А.Н. // Кинетика и катализ. 2010. Т. 51. № 3. С. 358.
Азатян В.В. // Там же. 1996. Т. 37. № 4. С. 537.
Прокопенко В.М., Азатян В.В. // Журн. физ. химии. 2018. Т. 92. № 1.
Азатян В.В., Бакулев В.И., Калканов В.А. и др. // Способ питания газового двигателя внутреннего сгорания. Пат. RUS 1835139, 1992.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии