

Журнал физической химии, 2021, T. 95, № 3, стр. 393-404
Формирование активных центров нанесенных на наноалмазы никель-цинковых катализаторов селективного гидрирования фенилацетилена
Е. В. Голубина a, *, Е. С. Локтева a, А. В. Ерохин a, В. Ю. Мурзин b, c, В. С. Черникова d, А. А. Велигжанин e
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b DESY
Hamburg, Germany
c University of Wuppertal
Wuppertal, Germany
d King Abdullah University of Science and Technology
Thuwal, Kingdom of Saudi Arabia
e Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: golubina@kge.msu.ru
Поступила в редакцию 01.08.2020
После доработки 01.08.2020
Принята к публикации 03.08.2020
Аннотация
Изучены биметаллические катализаторы NiZn/ND с соотношением Ni : Zn = 1 : 1 и 1 : 3, приготовленные методом пропитки; в качестве носителя использован детонационный наноалмаз. Проведено сравнение с монометаллическими Ni/ND и Zn/ND. Методами адсорбции/десорбции азота, сканирующей и просвечивающей электронной микроскопии показано, что нанесение металлов не влияет на пористую структуру и морфологию носителя. Координирование прекурсоров металлов на поверхности наноалмаза происходит с участием функциональных групп, что подтверждается изменением электрокинетического заряда поверхности. Восстановление прекурсоров металлов исследовано методами температурно-программированного восстановления и in situ XAFS-спектроскопии. В Ni-содержащих образцах обнаружены две формы Ni2+, по-разному связанные с носителем. В процессе обработки водородом до 400°С ZnO в образцах не восстанавливается. Доля восстановленного никеля определена на основании анализа XANES-спектров. В катализаторе с соотношением Ni : Zn = 1 : 1 в течение 4 ч in situ обработки водородом при 400°С в ячейке спектрометра происходит практически полное восстановление никеля, тогда как при соотношении Ni : Zn = 1 : 3 его полного восстановления в этих условиях не происходит. В реакции гидрирования фенилацетилена наибольшую селективность образования стирола во всем исследованном интервале температур 100–350°С обеспечивает NiZn/ND; NiZn3/ND менее активен и селективен, поскольку ZnO закрывает активные никелевые центры и препятствует адсорбции фенилацетилена.
Детонационные наноалмазы (ND) – перспективные системы для применения в катализе [1–6]. Уникальные свойства наноалмаза (высокая удельная площадь поверхности и присутствие большого числа функциональных групп) позволяют направленно влиять на формирование активных центров нанесенных на ND металлсодержащих катализаторов путем регулирования взаимодействия металл–носитель [2, 4, 7].
Селективное гидрирование тройной углерод-углеродной связи до двойной служит важной стадией очистки промышленного сырья от ацетиленовых примесей при полимеризации этилена, бутадиена и стирола, а также в синтезе малотоннажных химических продуктов [8–10]. Присутствие небольших примесей алкинов в сырье приводит к отравлению катализаторов полимеризации олефинов. Так, снижение количества фенилацетилена (ФА) в стирольном сырье позволяет существенно повысить качество продуктов полимеризации стирола, например, полистирола и пенополистирола [11].
Эффективными и селективными катализаторами реакции гидрирования алкинов служат нанесенные на углеродные и оксидные носители металлы или сплавы. Наиболее часто применяют системы на основе частиц Pd [12–16], но имеются также сведения об использовании платиновых катализаторов [17], сплавов Pd–Pt [16], Pd–Ag [18, 19], Ni–Pd, Fe–Pd, Mg–Pd [20]. Важной задачей является замена каталитических систем на основе благородных металлов более дешевыми, включающими другие переходные металлы, например, никель [21, 22]. Перспективные свойства проявляют катализаторы селективного гидрирования, в которых носителем активных компонентов (Ni, Cu и Pd) служил наноалмаз [2, 23, 24].
Оптимизация свойств катализаторов гидрирования тройной связи углерод–углерод основана на облегчении десорбции образующегося алкена с целью предотвращения его дальнейшего гидрирования. Расчетными и экспериментальными методами показано, что модификация Ni добавками Zn приводит к снижению энергии адсорбции этилена, в результате существенно возрастает выход этилена и снижается доля продуктов олигомеризации при гидрировании ацетилена [25, 26]. Сравнение различных модификаторов в реакции гидрирования фенилацетилена на биметаллических катализаторах Ni-M/AlSBA-15 показало, что наибольшая селективность по стиролу достигается при введении Zn или Ga [27]. Высокая эффективность достигалась благодаря формированию сплава никеля с цинком, а сравнение катализаторов с различными соотношениями металлов показало, что NiZn3/AlSBA-15 обеспечил 90.3% селективность образования стирола из фенилацетилена при близкой к 100% конверсии. Авторы [26] использовали метод теории функционала плотности для выявления наиболее селективного биметаллического Ni–Zn катализатора гидрирования ацетилена. Согласно расчетам, NiZn-катализатор с соотношением металлов 1 : 3 должен обладать наибольшей селективностью среди всех рассмотренных систем. Кроме того, добавление Zn способствует формированию более высокодисперсных частиц никеля, ограничивая их рост [28, 29].
В настоящей работе исследованы физико-химические свойства биметаллических NiZn-катализаторов, нанесенных на ND, с разным соотношением Ni : Zn и проведено сравнение с монометаллическими системами Ni/ND и Zn/ND. Каталитические свойства тестировали в реакции гидрирования фенилацетилена. Формирование активных центров в ходе координации предшественников активного компонента с функциональными группами на поверхности ND изучено с применением XAFS-спектроскопии с in situ восстановлением.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве носителя использовали детонационный наноалмаз (ЗАО “Синта”). Катализаторы готовили методом влажной пропитки: носитель пропитывали рассчитанным количеством водного раствора Ni(NO3)2⋅ 6H2O (“ч.д.а.”, “Реахим”, Москва) или Zn(NO3)2⋅ 6H2O (“ч.д.а.”, “Реахим”, Москва). Для приготовления биметаллических катализаторов проводили однократную пропитку раствором, содержащим нитраты обоих металлов. Затем образцы при перемешивании сушили сначала на водяной бане, а затем при 150°С в течение 2 ч. Восстановление проводили в токе водорода (12 мл/мин) при 380°С в течение 3 ч. Готовили биметаллические образцы NiZn/ND с двумя массовыми соотношениями металлов: в образце NiZn3/ND отношение Ni : Zn = 1 : 3 (1.25 мас. % Ni, 3.75 мас. % Zn), в образце NiZn/ND – 1 : 1 (2.5 мас. % Ni, 2.5 мас. % Zn). Для сравнения использовали два монометаллических катализатора 2.5 мас. % Ni/ND и 1.0 мас. % Zn/ND.
Содержание металлов контролировали методом атомно-абсорбционной спектроскопии на приборе Thermo iCE 3000 (Thermo Fisher Scientific Inc., США). Для перевода металлов в раствор образцы обрабатывали при 90°С в смеси HNO3/H2SO4 (соотношение масс 1 : 1).
Электрокинетический заряд поверхности образцов в суспензии в деионизованной воде определяли на приборе ZETASIZER nano-ZS (Malvern, Великобритания).
Изотермы адсорбции–десорбции азота регистрировали при 77 К на автоматизированном сорбтометре Autosorb-1 (Quantachtome, США). Удельную площадь поверхности и средний диаметр пор рассчитывали методами БЭТ и BJH соответственно.
Электронные микрофотографии поверхности образцов получали на сканирующем электронном микроскопе JEOL JSM-6390 (JEOL Ltd, Япония), оснащенном приставкой для локального энергодисперсионного анализа (СЭМ-ЭДА); снимки просвечивающей электронной микроскопии высокого разрешения (ПЭМ ВР) – на приборе JEM 2100F-UHR (JEOL Ltd, Япония) с приставкой для локального энергодисперсионного анализа (ЭДА).
Температурно-программированное восстановление невосстановленных образцов проводили на лабораторной установке при нагревании в токе 5%H2 в Ar. Нагревание осуществляли со скоростью 12 К/мин. Поглощение водорода определяли с помощью детектора по теплопроводности, обработку данных осуществляли с использованием программы “Экохром”.
XAFS-исследования проводили с использованием синхротронного излучения на станции “Структурное материаловедение” Курчатовского источника синхротронного излучения (НИЦ “Курчатовский институт”) [30]. Спектры XANES/EXAFS вблизи K-края поглощения Ni и Zn измеряли “по пропусканию” с использованием ионизационных камер, расположенных перед и после образца. Токовые сигналы с ионизационных камер измеряли цифровыми пикоамперметрами Keithley 6487. Сканирование по энергии осуществляли путем вращения кремниевого моноблочного монохроматора с прорезью, отражающие плоскости которого ориентированы по кристаллографическому направлению (111). Монохроматор обеспечивает энергетическое разрешение ΔЕ/Е = 2 × 10–4. Шаг по энергии составлял 0.5 эВ для спектров XANES и 1.5–2.5 эВ для спектров EXAFS. Для абсолютной калибровки энергетической шкалы использовали спектры стандартов (фольга соответствующего металла). Один спектр EXAFS измеряли за 20 мин.
Для in situ XAFS-исследования образец смешивали со связующим (гексагональным нитридом бора) и прессовали самоподдерживающуюся таблетку объемом ~100 мм3. Для нагрева и обработки газами использовали специализированную ячейку [31]. Для обработки образца использовали поток 400 мл/мин газовой смеси 2%H2 + 98%He, обеспечивающий газообмен в in situ ячейке. Нагрев проводили до температуры 400°С с шагом 100 К. Для каждой ступени нагрева образец выдерживали в изотермическом режиме и снимали несколько спектров. Обработку результатов проводили с использованием пакета программ IFEFFIT [32, 33]. Вейвлет-преобразование EXAFS-спектров осуществляли с использованием модифицированной функции Морле, как описано в [2].
Гидрирование фенилацетилена осуществляли импульсным методом в интервале температур 100–300°С в реакторе с неподвижным слоем катализатора, снабженном линией подачи H2 при постоянном атмосферном давлении и ловушкой для накопления продуктов, охлаждаемой ледяной водой. Водород подавали через верхнюю часть реактора. При каждой температуре реакции фенилацетилен впрыскивали с помощью шприца в поток водорода импульсами (0.23 ммоль, 3 импульса при заданной температуре с интервалами в 10 мин). После трех импульсов реактор выдерживали при заданной температуре в течение 20 мин. Затем продукты, собранные в ловушке, анализировали методом хромато-масс-спектрометрии на приборе Thermo Trace DSQ II (Thermo, США; колонка DB-5: 15 м, внутренний диаметр 0.25 мм). Тщательное сканирование масс-спектров показало, что стирол и этилбензол – единственные продукты реакции.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Физико-химические характеристики образцов и исследование методом электронной микроскопии
Результаты исследования методом адсорбции/десорбции азота показали, что величина площади удельной поверхности для всех образцов лежит в интервале от 290 до 330 м2/г, а диаметр пор составляет 9.6 нм. Таким образом, нанесение металлов, как по одному, так и совместно, не оказывает существенного влияния на удельную поверхность катализаторов и размер пор.
Морфологию поверхности и распределение металлов исследовали методом СЭМ-ЭДА (рис. 1а–1д). Из микрофотографий видно, что после нанесения металлов структура поверхности не изменяется. Соотношение Ni : Zn в катализаторах, определенное методом ЭДА, соответствует рассчитанным теоретически и определенным методом ААС. Анализ карт распределения элементов показал, что области повышенной локализации металлов отсутствуют. Равномерное распределение металлов на поверхности характерно для детонационных наноалмазов за счет участия О‑содержащих функциональных групп поверхности в координировании прекурсора металла [2, 4]. Участие функциональных групп в связывании металлов очевидно из значений электрокинетического потенциала (ζ-потенциала) суспензий носителя и катализаторов в деионизированной воде. Исходный ND имеет отрицательный заряд –4.3 мВ вследствие диссоциации поверхностных кислотных групп в воде. Карбоксильные группы, лактоны, фенольные и лактольные группы способствуют кислотному характеру углеродных материалов [34]. После нанесения никеля значение ζ-потенциала становится положительным, ~0.5 мВ. Изменение ζ‑потенциала от отрицательного до положительного значения указывает на изменение количества поверхностных карбоксильных групп. Для Zn-содержащих катализаторов ζ-потенциал составляет ~4–5 мВ, независимо от содержания Zn и присутствия Ni. Этот факт, скорее всего, связан с присутствием на поверхности частиц ZnO, которые имеют положительный ζ-потенциал в области рН до 10.3 [35]. В случае Zn-содержащих катализаторов ZnO оказывает основное влияние на суммарное значение ζ-потенциала. На основании этих данных можно предположить, что ZnO устойчив к восстановлению.
Рис. 1.
Микрофотографии СЭМ (а–в), микрофотографии СЭМ и соответствующие карты распределения элементов (г, д) и ПЭМ ВР (e).

На микрофотографиях ПЭМ высокого разрешения (рис. 1е) отчетливо видна структура носителя в виде сферических алмазных нанокристаллов размером 4–5 нм, покрытых оболочкой, включающих атомы углерода в разной гибридизации [36]. ПЭМ-исследование подтверждает результаты СЭМ-ЭДА об отсутствии в образцах крупных металлсодержащих частиц. Таким образом, можно утверждать, что размер нанесенных частиц металлов не превышает нескольких нанометров. Видимо, небольшие нанесенные частицы имеют низкий контраст с ND и не могут быть надежно выделены на микрофотографиях ПЭМ. Присутствие никеля и цинка в образцах катализаторов подтверждается методом локального энергодисперсионного анализа и ААС.
Приведенные данные позволяют заключить, что на поверхности наноалмаза формируются наноразмерные частицы, скорее всего Ni0 и ZnO. Образованию нанодиспергированных частиц никеля способствуют особенности координации соли никеля с участием функциональных групп на поверхности в воздушной среде, описанные в [2, 4]. Нанесение металлов не оказывает влияния на морфологию поверхности и текстурные характеристики носителя.
Температурно-программированное восстановление
Температурно-программированное восстановление водородом (ТПВ) – весьма информативный метод изучения взаимодействия металл–носитель и процесса образования активных центров в катализаторах. Температура восстановления предшественника металла, зарегистрированная в профиле ТПВ, чувствительна к незначительным изменениям таких характеристик, как размер частиц, химическое состояние, сила взаимодействия металл–носитель и другие. Профили ТПВ исходного наноалмаза и невосстановленных предшественников катализаторов представлены на рис. 2.
Рис. 2.
Профили ТПВ исходного наноалмаза и невосстановленных предшественников катализаторов Zn/ND, Ni/ND, NiZn3/ND и NiZn/ND.
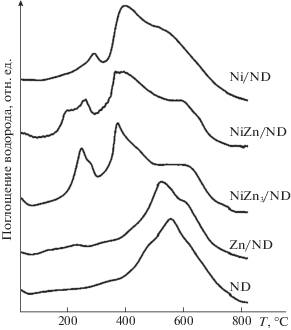
В профиле ТПВ исходного ND наблюдается широкий пик поглощения водорода при температуре выше 400°С. Этот пик соответствует гидрированию наноалмаза и присутствует в профилях ТПВ всех образцов. Профиль ТПВ невосстановленного Zn/ND существенно не отличается от профиля наноалмаза. Присутствие в образце ZnO приводит к небольшому смещению в область низких температур максимума пика поглощения водорода, относящегося к гидрированию ND. Имеется также малоинтенсивный пик при ~200°С и небольшое плечо при ~610°С. Общее количество поглощенного водорода в случае Zn/ND немного больше по сравнению с индивидуальным ND, но его недостаточно для полного восстановления всего ZnO, присутствующего в образце. В литературе отмечается, что ZnO стабилен в водороде при нагревании до 900°С [37, 38]. В то же время имеются сведения о возможности частичного восстановления ZnO с образованием ZnO1 – x в интервале температур от 400 до 650°С [39]. В [38] в условиях ТПВ наблюдали малоинтенсивные пики поглощения водорода при 200 и 500°C, которые авторы также связывают с частичным восстановлением ZnO. Таким образом, после восстановления в катализаторе Zn/ND можно ожидать присутствия в основном ZnO, хотя возможность частичного восстановления оксида цинка исключать нельзя. Полученный результат согласуется со значениями ζ‑потенциала Zn-содержащих образцов.
Профиль ТПВ невосстановленного образца Ni/ND содержит, помимо пика гидрирования носителя, два пика поглощения водорода при 260 и 390°C. Присутствие этих пиков связано с восстановлением двух форм никеля, по-разному связанных с наноалмазом [2]. Низкотемпературный пик (260°C) объясняется восстановлением физически адсорбированного NiO, слабо связанного с носителем. Высокотемпературный (390°C) пик восстановления обусловлен восстановлением частиц Ni2+, связанных с поверхностью ND вследствие химической координации предшественника никеля с O-содержащими функциональными группами на поверхности ND [2]. Сравнивая интенсивность пиков восстановления для Ni/ND с ранее полученными данными для 5% Ni/ND [2], можно отметить, что на профиле Ni/ND интенсивность низкотемпературного пика ниже, чем высокотемпературного, тогда как для 5% Ni/ND интенсивности обоих пиков сравнимы. Это свидетельствует о том, что большая часть Ni связана с функциональными группами поверхности наноалмаза, причем эти центры заполняются первыми, а оставшийся несвязанным с поверхностью прекурсор никеля формирует слабосвязанные формы.
Профили ТПВ обоих невосстановленных предшественников биметаллических катализаторов (рис. 2) также содержат две группы пиков восстановления NiO и один высокотемпературный широкий пик, соответствующий гидрированию носителя. При сравнении результатов ТПВ биметаллических образцов видно, что для NiZn/ND особенно заметно проявляется дополнительный низкотемпературный пик восстановления при 200°С. Появление дополнительных пиков с близкой температурой максимума может быть вызвано образованием смешанных оксидов никеля и цинка [40], температура восстановления которых будет отличаться от таковой для NiO. Еще одной причиной изменения температуры восстановления может быть активация водорода на поверхности ZnO с последующим восстановлением NiO при немного более низких температурах. Известно, что на поверхности оксида цинка уже при комнатной температуре может происходить диссоциативная адсорбция водорода [41, 42]. Поскольку пики в профилях ТПВ перекрываются, сложно провести точный расчет поглощения водорода в ходе восстановления разных частиц. На основании приближенного сравнения количества поглощенного водорода установлено, что в образце NiZn3/ND доля слабосвязанной формы никеля составляет ~30%. В составе NiZn/ND содержание этой формы меньше и составляет ~20%.
Таким образом, согласно данным ТПВ, модификация ZnO не оказывает кардинального влияния на процесс восстановления NiO, хотя никель и оксид цинка, вероятно, в некоторой степени контактируют в активном центре. Соотношение окисленных форм никеля зависит от соотношения Ni : Zn, причем при соотношении Ni : Zn = = 1 : 1 восстановление никеля начинает протекать при более низкой температуре, чем в составе образца с соотношением Ni : Zn = 1 : 3.
XAFS-исследование
Для определения возможного взаимодействия и степени контакта между соединениями никеля и цинка в активном компоненте проведено XAFS-исследование с использованием синхротронного излучения. Анализ протяженной тонкой структуры рентгеновского спектра поглощения (EXAFS-спектроскопия) использовали для получения сведений о локальной структуре и ближайшем окружении Ni и Zn. Исследовали процесс in situ восстановления катализаторов водородом, регистрируя спектры EXAFS и изменения в области околопороговой тонкой структуры спектров поглощения (XANES). Для более точной интерпретации полученных XAFS-данных также исследовали модельные соединения (нитрат цинка, ZnO, ацетат никеля и NiO).
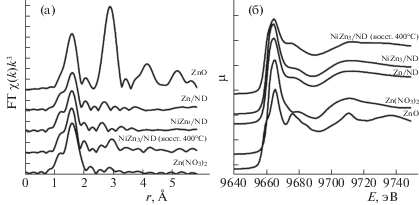
Кривые радиального распределения атомов (РРА), полученные фурье-преобразованием исходных данных, представлены на рис. 3а. Кривая РРА локального окружения цинка в ZnO аналогична описанным в литературе [43] для структуры вюрцита: первый пик на кривой РРА (r = 1.65 Å) соответствует расстоянию Zn–O в первой координационной сфере, а второй пик (r = 2.9 Å) соответствует Zn–O–Zn. Кривые РРА для катализаторов Zn/ND и NiZn3/ND совпадают. Максимум, наблюдаемый на удалении 1.69 Å от центрального атома, соответствует расстоянию Zn–O в оксиде цинка. Второй пик немного сдвинут относительно наблюдаемого на РРА массивного ZnO и соответствует r = 2.7 Å. Сдвиг и низкая интенсивность пика могут быть связаны с образованием мелких частиц ZnO в ходе нанесения на ND. Различия в положении максимумов, соответствующих расстояниям, превышающим 1.9 Å, наблюдаемые на РРА катализаторов и Zn(NO3)2, свидетельствуют об отсутствии нитрата цинка в невосстановленных прекурсорах катализаторов. Дополнительное восстановление NiZn3/ND in situ в ячейке EXAFS-спектрометра при 400°C привело к появлению пика на расстоянии r = 2.9 Å, что, скорее всего, связано с началом агрегации частиц ZnO. Восстановления цинка при такой обработке не происходит, поскольку первая координационная сфера Zn–O почти не изменяется. Анализ XANES K-края Zn (рис. 3б) также подтвердил, что оксидное состояние цинка остается стабильным при обработке в водороде при температуре 400°С в течение 5 ч. Наблюдается только упорядочение кристаллической решетки, что проявляется в более выраженных особенностях тонкой структуры спектра поглощения. Таким образом, полученные данные свидетельствуют о том, что после восстановительной обработки цинк находится в катализаторе в виде оксида.
Рис. 3.
Кривые радиального распределения атомов в локальном окружении Zn (а) и спектр XANES K-края поглощения Zn (б) для образцов Zn/ND, NiZn3/ND и модельных соединений.
Результаты XAFS для локального окружения никеля в катализаторах и модельных образцах представлены на рис. 4. На кривой РРА (рис. 4) для металлического никеля наблюдается максимум при r = 2.2 Å, соответствующий расстоянию Ni–Ni в первой координационной сфере. На кривой РРА чистого NiO присутствуют два пика на расстояниях 1.8 и 2.6 Å, соответствующие расстояниям Ni–O и Ni–O–Ni. Кривая РРА для NiZn3/ND представляет суперпозицию сигналов от Ni и NiO. При температуре обработки 280°С наблюдается расщепление пика, связанного с расстоянием Ni–O при R = 1.8 Å. Возникает компонента, соответствующая более короткому расстоянию Ni–O. Данное расстояние может быть отнесено к связям Ni–O, обусловленным координированием предшественника никеля с O-содержащими функциональными группами поверхности ND. Чтобы сравнить координацию никеля с карбоксильными группами на поверхности ND, в качестве модельного соединения выбрали ацетат никеля. На кривой РРА для ацетата никеля также наблюдается расщепление пика, отвечающего расстоянию Ni–O.
Рис. 4.
Кривые радиального распределения атомов в локальном окружении Ni для NiZn3/ND и модельных соединений (а); спектры XANES K-края Ni для Ni/ND, NiZn3/ND и NiO (б).
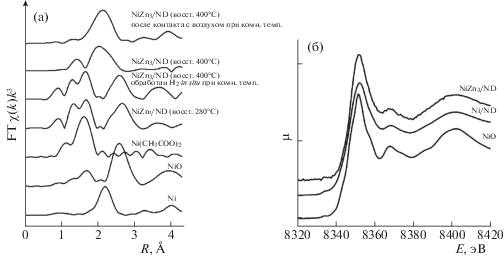
Таким образом, восстановленный катализатор содержит в своем составе Ni0 и NiO. Присутствие NiO в исходном образце может быть вызвано как неполным восстановлением оксида никеля, так и окислением частиц никеля, образовавшихся в ходе восстановления, при контакте с воздухом. Ранее установлено, что в составе восстановленного при 280°С катализатора 5% Ni/ND остается некоторое количество NiO [2]. Обработка водородом в ячейке прибора при комнатной температуре не приводит к изменениям РРА кривой NiZn3/ND. Повышение температуры восстановления до 400°С приводит к увеличению пика, соответствующего Ni0, что указывает на происходящее восстановление NiO. Форма кривой РРА восстановленного образца NiZn3/ND после контакта с воздухом существенно не изменяется, что свидетельствует об устойчивости образовавшихся частиц Ni0 к окислению.
Приведенные результаты показывают, что в отличие от цинка, координационное окружение никеля заметно изменяется в процессе восстановления. Более подробно восстановление никеля в биметаллических катализаторах NiZn3/ND и NiZn/ND исследовали в процессе in situ ступенчатого нагрева в восстановительной атмосфере в ячейке EXAFS-спектрометра. Об изменениях судили по спектрам XANES K-края Ni, а также дополнительно анализировали окружение никеля на основании спектра EXAFS. В качестве примера на рис. 5 приведены результаты для NiZn3/ND.
Рис. 5.
Спектры XANES K-края Ni (а) и кривые РРА в локальном окружении Ni для изотермической ступени 300°С (б) для NiZn3/ND в ходе in situ восстановления.
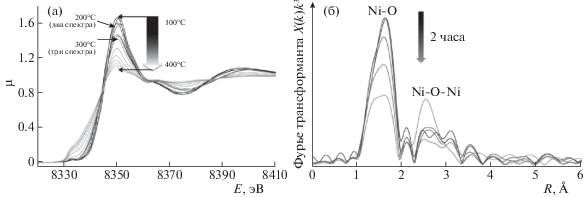
Вид XANES спектров K-края Ni (рис. 5а) в процессе обработки в восстановительной атмосфере заметно меняется и приближается к форме, характерной для металлического никеля. Оксид никеля начинает восстанавливаться при температуре 200–300°С, что соответствует данным ТПВ. Анализ спектров XANES и EXAFS позволил судить о процессах, происходящих с NiO в атмосфере водорода. В ходе ступенчатого повышения температуры происходит восстановление части NiO. Последующее выдерживание в изотермическом режиме сначала при температуре 200°С, а затем при 300°С не приводит к изменению формы K-края XANES-спектра, что свидетельствует о неизменности степени окисления. В то же время вид кривой РРА меняется. Так, при температуре 300°С (рис. 5б) хорошо видно уменьшение интенсивности пика, соответствующего первой координационной сфере, и одновременно увеличивается пик, отвечающий связи Ni–O–Ni второй координационной сферы. Таким образом, можно предположить, что перед восстановлением NiO до Ni0 протекает упорядочение структуры оксида никеля. Дальнейший нагрев и выдерживание при температуре 400°С приводят к постепенному повышению доли металлического никеля.
Степень восстановления NiO оценивали разложением спектра XANES на линейную комбинацию спектров для Ni0 и NiO. Изменение доли металлического никеля в процессе ступенчатого восстановления для биметаллических катализаторов представлено на рис. 6. Видно, что повышение температуры восстановления от 100 до 300°С, как уже отмечалось, сопровождается увеличением доли Ni0. Однако выдерживание в изотермическом режиме при каждой температуре не приводит к изменению окисленного состояния никеля. Сравнение данных для обоих биметаллических катализаторов показывает, что в исследованном температурном интервале доля Ni0 не зависит от соотношения Ni/Zn. А вот скорость восстановления при температуре 400°С зависит от этого соотношения. В ходе восстановления NiZn3/ND происходит медленное восстановление NiO, и через 6 ч доля Ni0 составила 73%. В катализаторе с соотношением Ni : Zn = 1 : 1 уже практически через 3 ч произошло весьма значительное восстановление NiO. После 4 ч обработки водородом при 400оС доля Ni0 составила 93%. Учитывая, что в результате восстановления происходит образование нанокристаллического никеля, а при расчете степени восстановления использовали данные для массивного кристаллического никеля, есть основания полагать, что в образце NiZn/ND восстановление NiO произошло полностью. Известно, что спектры XANES массивного и нанокристаллического Ni могут отличаться [44, 45].
Рис. 6.
Изменение доли металлического никеля в процессе восстановления водородом при ступенчатом повышении температуры для NiZn/ND и NiZn3/ND (а); вейвлет-трансформант EXAFS-спектра последней точки in situ исследования для NiZn/ND (б) и NiZn3/ND (в).
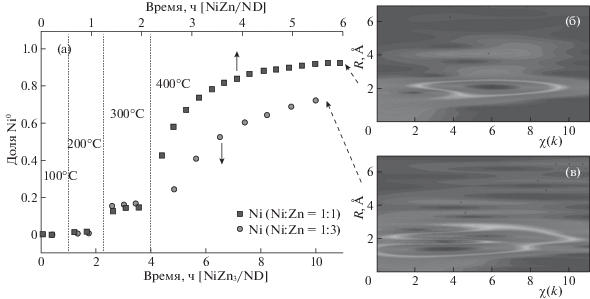
Полнота восстановления никеля в составе NiZn/ND также подтверждается результатами, полученными при обработке EXAFS-спектра с помощью вейвлет- преобразования с использованием модифицированной функции Морле [46]. Вейвлет-преобразование EXAFS-сигнала позволяет разделить вклады в рассеяние от атомов, находящихся на одинаковом расстоянии от центрального атома, но различающихся по атомной массе. Получающаяся трехмерная карта в пространственных координатах и координатах волновых чисел дает возможность провести более подробный анализ экспериментальных данных. Надежность использованного подхода проверяли путем сравнения экспериментальных данных для исследуемых образцов и модельных соединений. Результаты такой обработки представлены на рис. 6б и 6в в виде изображения вейвлет-трансформанта EXAFS-спектра, характеризующего зависимость амплитуды XAFS-функции при заданных волновом векторе (k) и расстоянии (R).
На полученном графике разложения результата фурье-преобразования сигнала EXAFS NiZn/ND, восстановленного при 400°С (рис. 6б), наблюдается один максимум, который связан с рассеиванием по пути Ni–Ni. Координаты этого максимума полностью совпадают с полученным результатом для модельного стандартного образца металлического Ni. Для образца NiZn3/ND (рис. 6в) на изображении вейвлет-трансформанта EXAFS-спектра присутствуют два пика. Максимум, соответствующий расстоянию ~2 Å и k ∼ 6, относится к Ni0. Второй максимум, характеризующийся меньшими значениями расстояния и k, связан с присутствием в образце NiO.
Таким образом, исследование процессов восстановления катализаторов показало, что ZnO не восстанавливается при обработке водородом при 400оС, а происходит лишь упорядочение кристаллической структуры оксида. Доля Ni0 в образцах зависит от соотношения металлов. В образце NiZn/ND в результате обработки водородом при 400°С за 4 ч происходит практически полное восстановление NiO до Ni0. Высокое содержание цинка в образце NiZn3/ND замедляет процесс, и полного восстановления NiO не происходит.
Каталитическая активность в гидрировании фенилацетилена
Каталитическую активность приготовленных в работе катализаторов сравнивали в реакции парофазного гидрирования фенилацетилена (ФА). В присутствии NiZn3/ND и Zn/ND конверсия ФА не превышала 30% (рис. 7а) во всем исследованном температурном интервале. При этом селективность образования стирола возрастала от 50–60 до 90% при повышении температуры реакции от 100 до 300°С. Низкая конверсия ФА в присутствии Zn/ND вполне закономерна, поскольку оксид цинка не обладает значительной каталитической активностью в реакции гидрирования. Наибольшая конверсия ФА достигнута в присутствии Ni/ND и NiZn/ND катализаторов (рис. 7а), однако ход температурной зависимости конверсии для этих двух образцов различался. В присутствии Ni/ND в интервале температур от 150 до 300°С наблюдается постепенное повышение конверсии до 98.5%, а в случае NiZn/ND в узком температурном диапазоне происходит резкое повышение конверсии ФА от 5% при 200°С до 94% при 250°С. Для обоих катализаторов приближение к полному превращению ФА сопровождается снижением селективности образования стирола до 50–60% (рис. 7б).
Рис. 7.
Температурные зависимости конверсии фенилацетилена (а) и селективности образования стирола (б) в присутствии моно- и биметаллических катализаторов.
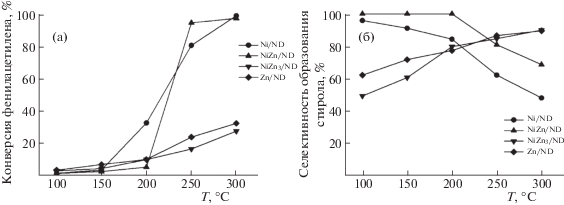
Гидрирование ФА на поверхности гетерогенных катализаторов протекает по механизму последовательно-параллельной реакции [12, 17]. Первый путь включает гидрирование ФА с последовательным образованием стирола и этилбензола. Второй путь реакции заключается в прямом гидрировании ФА до этилбензола. В соответствии с этой схемой реакции, повышению эффективности катализатора способствуют снижение вклада стадии прямого гидрирования ФА и облегчение десорбции стирола. Последнее наиболее эффективно достигается изменением адсорбционных характеристик катализатора.
Различия в каталитических свойствах биметаллических Ni–Zn-катализаторов на наноалмазе могут быть обусловлены возникновением новых свойств поверхности на границе контакта Ni–ZnO, а также изменением адсорбционных характеристик катализатора. Мы ожидали, что эффективность катализатора с соотношением Ni : Zn = 1 : 3 будет высокой, поскольку в литературе [26] отмечено, что именно это соотношение в NiZn-сплаве наиболее эффективно для достижения высокой активности и селективности по этилену при гидрировании ацетилена. Однако, как показано выше, на поверхности исследованных в нашей работе катализаторов цинк сохраняется в виде оксида, и биметаллического сплава не образуется. В то же время ZnO может изменять адсорбционные и каталитические свойства поверхности. Так, в присутствии ZnO может протекать дополнительная активация водорода. На основании результатов ТПВ установлено, то при соотношении Ni : Zn = 1 : 1 восстановление NiO начинается при более низких температурах. Поскольку метод EXAFS показал, что в невосстановленном образце присутствует только оксид никеля, снижение температуры его восстановления может быть связано с активацией водорода на частицах ZnO. При более высоком содержании цинка (Ni : Zn = 1 : 3) возможно декорирование поверхности частиц NiO оксидом цинка. Это приводит к затруднению восстановления NiO, что и наблюдается по результатам in situ XANES-EXAFS-исследования. В результате каталитическая активность образца NiZn3/ND примерно соответствует таковой для Zn/ND, т.е. определяется преимущественно каталитическими свойствами ZnO. Аналогичное объяснение предложено в литературе для описания низкой селективности образования этилена в ходе гидрирования ацетилена в присутствии PdZn/C-катализаторов: поверхность палладиевого активного центра блокируется ZnO [47].
Высокая селективность образования стирола в присутствии NiZn/ND может быть связана с достижением более низкой энергии адсорбции стирола на его поверхности по сравнению с монометаллическим Ni/ND. Также дополнительное повышение эффективности может происходить за счет эффектов переноса электрона на границе контакта Ni–ZnO. Так, ранее показано, что для повышения активности Ni–ZnO/SiO2 в реакции гидрирования ключевую роль играет перенос электрона с образованием Niδ+ и (ZnO)δ– [48]. Важную роль переноса электрона отмечали и в случае PdCu/Al2O3- и PdZn/Al2O3-катализаторов реакции гидрирования фенилацетилена [49]. По мнению авторов, перенос заряда от Pd к Zn оказывает сильное влияние на адсорбцию алкенов, облегчая десорбцию стирола. Схожий механизм может реализовываться и в системе Ni–Zn. В результате модифицирования Ni/ND оксидом цинка при соотношении Ni : Zn = 1 : 1 наблюдается увеличение селективности образования стирола во всем исследованном температурном интервале.
ЗАКЛЮЧЕНИЕ
Каталитические свойства биметаллических никель-цинковых катализаторов, нанесенных на детонационный наноалмаз, в реакции гидрирования фенилацетилена зависят от соотношения металлов. Относительно высокую селективность образования стирола проявили образцы NiZn3/ND и Zn/ND, однако конверсия ФА не превышала 30%. Катализатор NiZn/ND существенно превышает по активности другие содержащие цинк системы; он несколько более активен по сравнению с Ni/ND, а селективность по стиролу превышает активность Ni/ND во всем исследованном температурном интервале.
Установлено, что в приготовленных системах не наблюдается образования сплавов никеля с цинком. Координирование прекурсоров металлов на поверхности наноалмаза происходит с участием функциональных групп поверхности, что подтверждается изменением электрокинетического заряда поверхности. Присутствие ZnO вносит существенный вклад в значение ζ-потенциала. Методами ТПВ и XAFS показано, что ZnO не восстанавливается при обработке водородом при 400°С, происходит лишь упорядочение кристаллической структуры оксида, а доля восстановленного никеля в образцах зависит от соотношения Ni/Zn. В образце NiZn/ND в результате обработки водородом при 400°С за 4 ч происходит практически полное восстановление NiO до Ni0, чего не происходит в образце NiZn3/ND. При более высоком содержании цинка (Ni : Zn = 1 : 3) возможно декорирование поверхности частиц NiO оксидом цинка, затрудняющее восстановление NiO. Различия в каталитическом действии обусловлены совместным действием трех факторов: содержанием Ni0, адсорбционными свойствами и степенью переноса заряда от никеля к оксиду цинка.
Список литературы
Gupta N., Wang Q., Wen G., Su D. in: Nanodiamonds for Catalytic Reactions/Ed by J.-C. Arnault. Ch.18. Elsevier, 2017, p. 439. https://doi.org/10.1016/B978-0-32-343029-6.00019-2
Golubina E.V., Lokteva E.S., Erokhin A.V. et al. // J. Catalysis. 2016. V. 344. P. 90.https://doi.org/10.1016/j.jcat.2016.08.017
Vershinin N.N., Efimov O.N., Bakaev V.A. et al. // Fullerenes, Nanotubes and Carbon Nanostructures. 2010. V. 19. P. 63.https://doi.org/10.1080/1536383x.2010.490143
Mavrodinova V., Popova M., Kolev I. et al. // Applied Surface Science. 2007. V. 253. P. 7115.https://doi.org/10.1016/j.apsusc.2007.02.090
Tsoncheva T., Mavrodinova V., Ivanova L. et al. // J. Mol. Catalysis. A: Chemical. 2006. V. 259. P. 223.https://doi.org/10.1016/j.molcata.2006.06.019
Локтева Е.С., Голубина Е.В., Качевский С.А. и др. // Кинетика и катализ. 2011. V. 52. P. 149. [Lokteva E.S., Golubina E.V., Kachevskii S.A. et al. // Kinetics and Catalysis. 2011. V. 52. P. 145 (DОI: 10.1134/S0023158411010125)]
Lokteva E.S., Golubina E.V. // Pure and Appl. Chem. 2019. V. 91. P. 609.https://doi.org/10.1515/pac-2018-0715
Lonergan W.W., Vlachos D.G., Chen J.G. // J. Catalysis. 2010. V. 271. P. 239.https://doi.org/10.1016/j.jcat.2010.01.019
Borodziński A., Bond G.C. // Catalysis Reviews. 2008. V. 50. P. 379.https://doi.org/10.1080/01614940802142102
Huang W., McCormick J.R., Lobo R.F., Chen J.G. // J. Catalysis. 2007. V. 246. P. 40.https://doi.org/10.1016/j.jcat.2006.11.013
Басимова Р.А., Павлов М.Л. и др. // Нефтехимия. 2009. V. 49. P. 380 [Basimova R.A., Pavlov M.L., Myachin S.I. et al. // Petroleum Chemistry. 2009. V. 49. № 5. P. 360. https://doi.org/10.1134/S096554410905003X
Deng D., Yang Y., Gong Y. et al. // Green Chemistry. 2013. V. 15. P. 2525.https://doi.org/10.1039/c3gc40779a
Dominguez-Dominguez S., Berenguer-Murcia A., Linares-Solano A., Cazorla-Amoros D. // J. Catalysis. 2008. V. 257. P. 87.https://doi.org/10.1016/j.jcat.2008.04.008
Markov P.V., Mashkovsky I.S., Bragina G.O. et al. // Chemical Engineering Journal. 2019. V. 358. P. 520.https://doi.org/10.1016/j.cej.2018.10.016
Weerachawanasak P., Mekasuwandumrong O., Arai M. et al. // J. Catalysis. 2009. V. 262. P. 199.https://doi.org/10.1016/j.jcat.2008.12.011
Carturan G., Cocco G., Facchin G., Navazio G. // J. Mol. Catalysis. 1984. V. 26. P. 375.https://doi.org/10.1016/0304-5102(84)85111-1
Wilhite B.A., McCready M.J., Varma A. // Industrial & Engineering Chemistry Research. 2002. V. 41. P. 3345.https://doi.org/10.1021/ie0201112
Huang W., Pyrz W., Lobo R.F., Chen J.G. // Applied Catalysis A: General. 2007. V. 333. P. 254.https://doi.org/10.1016/j.apcata.2007.09.017
He Y., Liu Y., Yang P. et al. // J. Catalysis. 2015. V. 330. P. 61.https://doi.org/10.1016/j.jcat.2015.06.017
Dominguez-Dominguez S., Berenguer-Murcia A., Cazorla-Amoros D., Linares-Solano A. // J. Catalysis. 2006. V. 243. P. 74.https://doi.org/10.1016/j.jcat.2006.06.027
Donphai W., Kamegawa T., Chareonpanich M., Yamashita H. // Industrial & Engineering Chemistry Research. 2014. V. 53. P. 10105.https://doi.org/10.1021/ie5014597
Chen X., Zhao A., Shao Z. et al. // J. Phys. Chem. C. 2010. V. 114. P. 16525.https://doi.org/10.1021/jp1050832
Huang F., Jia Z., Diao J. et al. // J. Energy Chem. 2019. V. 33. P. 31.https://doi.org/10.1016/j.jechem.2018.08.006
Sun Y., Luo B., Xu S. et al. // Chem. Phys. Letters. 2019. V. 723. P. 39.https://doi.org/10.1016/j.cplett.2019.03.015
Spanjers C.S., Held J.T., Jones M.J. et al. // J. Catalysis. 2014. V. 316. P. 164.https://doi.org/10.1016/j.jcat.2014.05.007
Studt F., Abild-Pedersen F., Bligaard T. et al. // Sci. 2008. V. 320. P. 1320.https://doi.org/10.1126/science.1156660
Yang L., Yu S., Peng C. et al. // J. Catalysis. 2019. V. 370. P. 310.https://doi.org/10.1016/j.jcat.2019.01.012
Takahashi H., Sunagawa Y., Myagmarjav S. et al. // Materials Transactions. 2003. V. 44. P. 2414.https://doi.org/10.2320/matertrans.44.2414
Myagmarjav S., Takahashi H., Sunagawa Y. et al. // MATERIALS TRANSACTIONS. 2004. V. 45. P. 2035.https://doi.org/10.2320/matertrans.45.2035
Chernyshov A.A., Veligzhanin A.A., Zubavichus Y.V. // Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 2009. V. 603. P. 95.https://doi.org/10.1016/j.nima.2008.12.167
Veligzhanin A.A., Zubavichus Y.V., Chernyshov A.A. et al. // J. Struct.Chem. 2010. V. 51. P. 20.https://doi.org/10.1007/s10947-010-0186-9
Ravel B., Newville M. // Journal of Synchrotron Radiation. 2005. V. 12. P. 537.https://doi.org/10.1107/S0909049505012719
Newville M. // J. Synchrotron Radiation. 2001. V. 8. P. 322.https://doi.org/10.1107/S0909049500016964
Gibson N., Shenderova O., Luo T.J. M. et al. // Diamond and Related Materials. 2009. V. 18. P. 620.https://doi.org/10.1016/j.diamond.2008.10.049
Marsalek R. // APCBEE Procedia. 2014. V. 9. P. 13.https://doi.org/10.1016/j.apcbee.2014.01.003
Krueger A. // J. Materials Chem. 2008. V. 18. P. 1485.https://doi.org/10.1039/b716673g
McCluskey M.D., Jokela S.J., Hlaing O.W.M. // Physica B: Condensed Matter. 2006. V. 376–377. P. 690.https://doi.org/10.1016/j.physb.2005.12.173
Chin Y.-H., Dagle R., Hu J. et al. // Catalysis Today. 2002. V. 77. P. 79.https://doi.org/10.1016/S0920-5861(02)00234-1
Wang W., Li X., Zhang Y. et al. // Catalysis Science & Technology. 2017. V. 7. P. 4413.https://doi.org/10.1039/C7CY01119A
Baranov A.N., Sokolov P.S., Kurakevych O.O. et al. // High Pressure Research. 2008. V. 28. P. 515.https://doi.org/10.1080/08957950802379307
Conner W.C., Kokes R.J. // J. Catalysis. 1975. V. 36. P. 199.https://doi.org/10.1016/0021-9517(75)90024-X
Anderson A.B., Nichols J.A. // J. Amer. Chem. Soc. 1986. V. 108. P. 4742.https://doi.org/10.1021/ja00276a010
Timoshenko J., Anspoks A., Kalinko A., Kuzmin A. // Physica Status Solidi (C). 2014. V. 11. P. 1472.https://doi.org/10.1002/pssc.201300615
Koningsberger D.C., Ramaker D.E., Applications of X‑ray Absorption Spectroscopy in Heterogeneous Catalysis: EXAFS, Atomic XAFS, and Delta XANES, in: Handbook of Heterogeneous Catalysis, 2008, p. 774. https://doi.org/10.1002/9783527610049.hetcat0040
Iwasawa Y. X-Ray Absorption Fine Structure for Catalysts and Surfaces. in: Series on Synchrotron Radiation Techniques and Applications, Singapore: World Scientific Publishing, 1996. 410 p. https://doi.org/10.1142/2807
Timoshenko J., Kuzmin A. // Computer Physics Comm. 2009. V. 180. P. 920https://doi.org/10.1016/j.cpc.2008.12.020
Glyzdova D.V., Afonasenko T.N., Khramov E.V. et al. // Topics in Catalysis. 2020. V. 63. P. 139.https://doi.org/10.1007/s11244-019-01215-9
Wang J., Jin H., Wang W.-H. et al. // ACS Applied Materials & Interfaces. 2020. V. 12. P. 19581.https://doi.org/10.1021/acsami.0c03037
Wang Z., Yang L., Zhang R. et al. // Catalysis Today. 2016. V. 264. P. 37.https://doi.org/10.1016/j.cattod.2015.08.018
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии