

Журнал физической химии, 2021, T. 95, № 3, стр. 374-392
Селективное каталитическое восстановление оксидов азота углеводородами в избытке кислорода
В. А. Садыков a, b, *, В. А. Матышак c
a Российская академия наук, Сибирское отделение, Институт катализа им. Г.К. Борескова
Новосибирск, Россия
b Новосибирский государственный университет
Новосибирск, Россия
c Российская академия наук, Институт химической физики им. Н.Н. Семенова
Москва, Россия
* E-mail: vasadykov@mail.ru
Поступила в редакцию 06.08.2020
После доработки 06.08.2020
Принята к публикации 10.09.2020
Аннотация
В данном обзоре представлен анализ результатов работ, проведенных с активным участием В.В. Лунина в области селективного каталитического восстановления оксидов азота углеводородами в избытке кислорода. С помощью уникальной совокупности спектральных и кинетических методов установлены основные характеристики механизма реакций, включающего ключевую стадию взаимодействия нитрит-нитратных комплексов с углеводородами. Таким образом создана основа для решения проблемы дизайна активных компонентов катализаторов данных процессов, обеспечивающих высокие покрытия нитрит-нитратных комплексов в условиях реального катализа, высокие скорости активации углеводородов на таких центрах и их эффективное взаимодействие с нитрит-нитратными комплексами. Это было достигнуто путем изменения типа катиона переходного металла на поверхности носителя, его ближайшего координационного окружения и степени кластерирования, а также основности носителя. Для активных компонентов на основе катионов меди, кобальта, никеля и железа и таких носителей как высококремнистые цеолиты, частично стабилизированный диоксид циркония, пилларированные диоксидом циркония природные глины и каркасные фосфаты циркония, в том числе промотированные серебром или платиной, такой подход позволил обеспечить высокую активность, селективность и стабильность в целевых реакциях в реальных условиях. Для практического использования этих активных компонентов были разработаны методы их нанесения на блочные носители сотовой структуры или формовки в виде блоков с использованием связующих, что создало эффективные катализаторы, защищенные патентами РФ.
ВВЕДЕНИЕ
Научно-исследовательские и опытно-конструкторские работы, направленные на создание новых каталитических процессов удаления оксидов азота из отходящих газов автотранспорта и промышленных предприятий, способных заменить дорогостоящий процесс селективного каталитического восстановления оксидов азота аммиаком, имеют большое значение для защиты окружающей среды. Для выбросов с большим избытком кислорода, такие как отходящие газы дизельных двигателей, ТЭЦ и др., привлекательной альтернативой может быть процесс селективного каталитического восстановления оксидов азота углеводородами (СКВ-УВ), независимо открытый Ивамото [1, 2] и Хелдом [3, 4]. К настоящему времени известны сотни публикаций и патентов по данным реакциям, которые частично обобщены в ряде обзоров [5–11]. В настоящем обзоре представлен анализ результатов совместных работ по данному направлению с начала 90-х годов 20-го века Института катализа СО РАН, Института химической физики, Института нефтехимического синтеза и Химического факультета МГУ с активным участием В.В. Лунина и сотрудников его лаборатории, которые внесли огромный вклад в исследования, в том числе, в рамках международного сотрудничества с европейскими учеными (Джулиан Росс из Ирландии и др.) [12–65 ]. При разработке новых катализаторов использовался системный иерархический подход, базирующийся на установлении основных закономерностей механизма этих реакций и природы активных центров поверхности для базовых типов катализаторов (катион-замещенных цеолитов, нанесенных оксидных систем, в том числе промотированных металлами платиновой группы, столбчатых глин), с использованием полученной информации для оптимизации состава и структуры активного компонента. Заключительный этап был связан с разработкой основ технологии приготовления блочных катализаторов для данных процессов.
Детальные, в том числе in situ, исследования механизма реакций с использованием в качестве восстановителей как парафинов, так и олефинов показали [12–23, 29, 40, 55, 56], что для систем, содержащих катионы переходных металлов, которые позволяют обеспечить высокую эффективность удаления оксидов азота при больших нагрузках в реальных условиях выбросов, ключевыми интермедиатами являются прочносвязанные нитрит-нитратные комплексы, локализованные на координационно-ненасыщенных центрах поверхности. Кроме того, была показана важность стадии активации углеводородов, протекающих как с непосредственным участием нитрит-нитратных комплексов, так и без их участия. Показано, что комбинация некоторых катионов переходных металлов с платиной или серебром позволяет обеспечить сопряжение между стадиями активации оксидов азота и углеводородов, что обеспечивает эффективное образование нитроорганических соединений – интермедиатов, содержащих азот, углерод и кислород. Превращение этих интермедиатов через цепочку последовательных и параллельных стадий, в том числе с участием кислотных центров поверхности, хотя и не лимитирует процесс, однако, влияет на селективность образования нежелательного побочного продукта – закиси азота [22]. Данные результаты позволили сформулировать основные требования к свойствам активных центров, включая их пространственное распределение на носителе.
В исследованиях большое внимание было уделено активным компонентам на основе катион-замещенных цеолитов со структурой типа ZSM-5 и нанесенным на оксид алюминия катионам переходных металлов – меди и кобальта [12–26, 58]. Это позволило выявить роль кластерирования катионов переходных металлов – меди и кобальта на таких носителях, кислотности носителей, а также выработать подходы к стабилизации кластеров в структуре цеолита и самого цеолита в гидротермальных условиях путем введения щелочноземельных катионов [27, 28].
Несмотря на определенные успехи в стабилизации структуры и каталитической активности цеолитсодержащих катализаторов для рассматриваемых процессов, термодинамическая нестабильность структуры цеолита неизбежно приводит к ограниченному сроку службы таких систем вследствие их деалюминирования, особенно если в процессе эксплуатации возможны перегревы. Поиск новых систем, обладающих стабильностью в условиях реальных выбросов, содержащих воду и диоксид серы, позволил остановиться на системах, содержащих катионы циркония, как наиболее перспективных для создания практических катализаторов. Такие системы включают в себя мезопористые катализаторы на основе частично стабилизированного диоксида циркония [29–37, 53], ультрамикропористые глины, пилларированные наночастицами диоксида циркония [36–49, 55, 57, 59, 61], и комплексные каркасные фосфаты циркония, с нанесенными или внедренными в каркас катионами переходных металлов [50–52]. Были разработаны основные подходы к синтезу таких систем с заданными пористой структурой, текстурой, фазовым составом и свойствами центров поверхности с использованием методов золь–гель-синтеза и механической активации смеси твердых солей с последующей гидротермальной обработкой, в том числе в присутствии ПАВ, а также к пилларированию природных алюмосиликатов – глин полиядерными гидроксокомплексами циркония контролируемого размера. Генезис этих систем был изучен с помощью комплекса современных структурных и спектральных методов.
Для этих перспективных систем были разработаны процедуры нанесения наноструктурированных активных компонентов, состоящих из катионов переходных металлов (их кластеров) с фиксированными на них кластерами металлов платиновой группы, и свойства таких компонентов были детально охарактеризованы. Оптимальные катализаторы были сформованы в виде блоков сотовой структуры или нанесены на различные блочные носители. В реалистических условиях, при нагрузках до 100 тыс. обратных часов, при использовании в качестве восстановителей пропана, пропилена или декана, эти катализаторы обеспечивают удаление до 80% оксидов азота без заметного образования закиси азота и без потери активности во времени. Эти катализаторы защищены рядом российских патентов [27, 28, 37, 48, 49].
В обзоре будут представлены основные аспекты данных работ с акцентом на наиболее значимые и оригинальные моменты.
1. ОСНОВНЫЕ ДЕТАЛИ МЕХАНИЗМА РЕАКЦИИ СЕЛЕКТИВНОГО КАТАЛИТИЧЕСКОГО ВОССТАНОВЛЕНИЯ ОКСИДОВ АЗОТА УГЛЕВОДОРОДАМИ В ИЗБЫТКЕ КИСЛОРОДА
1.1. Поверхностные формы оксидов азота
Установлено, что при адсорбции NO(NO + O2) на поверхности катализаторов образуются несколько типов поверхностных соединений, такие как нитрозилы, нитриты и нитраты. По данным ТПД и ИК-экспериментов нитрозилы являются слабосвязанными и десорбируются с поверхности при температурах порядка 100–200°С. Наблюдаемый в этой области пик выделения NO2 относится к разложению нитритных комплексов. Высокотемпературный (400–500°С) пик выделения NO отнесен к разложению прочно связанных поверхностных комплексов, таких как нитраты. Так же, как и в случае низкотемпературного пика NO2, высокотемпературный пик NO2 отнесен к разложению более прочносвязанных нитритных комплексов. С ростом содержания введенных в носитель (цеолит, столбчатая глина, оксид алюминия или циркония) катионов металла доля нитратных комплексов увеличивается (до 1% монослоя), а нитритных уменьшается. Следовательно, при температурах реального катализа ~300–400°С поверхностная концентрация нитрозильных комплексов мала, и комплексы этого типа не могут принимать значительного участия в реакции СКВ. Из этого следует, что интермедиатами реакции СКВ на образцах с высоким содержанием активного компонента являются прочносвязанные нитратные комплексы, но в других случаях возможно участие и нитритных комплексов.
На основании данных ИК-спектроскопии с использованием изотопзамещеного оксида азота 15N18O и теоретических расчетов была предложена структура нитратных комплексов, которые являются бидентатными с изолированным NO-фрагментом (табл. 1).
Таблица 1.
| Система | Положение полосы поглощения, см–1 | Изотопный сдвиг*, см–1 | Отнесение |
|---|---|---|---|
| CuZSM-5 Si/Al = 25, 2.8% Cu [15] |
1630 | 80 | мостиковый или бидентатный нитрат |
| 1570 | 70 | то же | |
| 1510 | 60 | нитрит | |
| 1244, 1279 (weak) | – | нитрит и/или нитрат | |
| CoZSM-5 Si/Al = 12, 2.3% Co [15] |
1610 | 60 | нитрит |
| 1600 | 50 | нитрит | |
| 1575, 1450 | 70 | мостиковый или бидентатный нитрат | |
| 1353, 1265 (weak) | – | нитрит или нитрат | |
| Дисперсный ZrO2 (прокален при 500°C, тетрагональная фаза) [29] |
1600–1612 | – | мостиковый нитрат |
| 1590–1584 1562–1556 |
– – |
бидентатный нитрат монодентатный нитрат |
|
| 1290–1244 | – | нитрат | |
| 1300–1360 | – | нитрит | |
| 1190 | – | нитрит | |
| Zr-PILC [62] | 1617 1257 |
– | мостиковый нитрат |
| 1588 1248 |
– | бидентатный нитрат |
Необходимо отметить отсутствие монодентатных нитратов для наночастиц диоксида циркония в столбчатых глинах, состоящих из тетрамеров Zr4(ОН)8 и их димеров Zr8(ОН)m что, очевидно, связано со спецификой их структуры, в частности, доминированием мостиковых гидроксильных групп и сильным взаимодействием с алюмосиликатными слоями [59].
1.2. Реакционная способность и маршруты трансформации нитрит-нитратных комплексов
Поскольку для установления механизма каталитической реакции ключевым критерием является сопоставление скорости каталитической реакции со скоростью образования продукта из потенциального интермедиата в идентичных условиях [64, 65], в наших исследованиях для широкого круга катализаторов было проведено систематическое сопоставление стационарных скоростей реакций селективного восстановления оксидов азота различными углеводородами со скоростями превращения нитратных комплексов под воздействием углеводородов в смеси с кислородом или без него (с использованием ИК-фурье-спектроскопии и вакуумных или проточных ячеек) и скоростями образования целевого продукта – азота под воздействием импульса смеси, содержащей углеводород, в том числе с кислородом (проточные микрокаталитические установки) [12–26, 29, 33, 38, 61–65]. Во всех случаях скорость превращения поверхностных нитратов хорошо описывалась уравнением первого порядка по их концентрации. Комбинация полученных констант скоростей поверхностной трансформации нитратов с данными по поверхностной концентрации, полученными методом ТПД для этих образцов, позволила рассчитать максимальную скорость расходования поверхностных нитратов, нормированную на выделение молекулярного азота при этих условиях. В качестве примера в табл. 2 приведены значения скоростей выделения продукта – молекулярного азота, рассчитанные из данных различных экспериментов для катализаторов на основе цеолита ZSM-5 при использовании в качестве восстановителя пропана [15, 23]. Близость скоростей превращения нитратных комплексов и скоростей каталитической реакции показывает, что нитратные комплексы действительно являются ключевыми интермедиатами. Причем активность катализатора Cu-ZSM-5 практически не изменяется при введении кислорода в смесь для титрования, в то время как катализатор Co-ZSM-5 при использовании смеси, содержащей только пропан, показал низкую активность, скорость выделения азота составила ~0.1 × × 1013 мол. NO/(м2 с), а при использовании смеси содержащей кислород скорость оказалась в несколько раз выше. Это факт можно объяснить тем, что при отсутствии кислорода в газовой фазе часть наиболее реакционноспособных поверхностных комплексов реагирует с пропаном давая NO, а при наличии кислорода более быстрым является маршрут трансформации с образованием молекулярного азота. Наиболее сильно это выражено для системы Co-ZSM-5.
Таблица 2.
Скорости выделения азота (молек./(м2 с)) под воздействием смесей с пропаном, рассчитанные из разных данных в стационарных и нестационарных условиях. Смесь 0.1% NO + 0.1% C3H8 + 1% O2 в He, 210°С
| Образец | ${{{v}}_{1}}$ × 10–13 | ${{{v}}_{2}}$ × 10–13 | ${{{v}}_{3}}$ × 10–13 | ${{{v}}_{4}}$ × 10–13 |
|---|---|---|---|---|
| Сu/ZSM-5 | 0.5 | 0.5 | 1.8 | 1.8 |
| Co/ZSM-5 | 0.3 | 1 | 0.8 | <0.1 |
Для катионзамещенных цеолитов титрование нитратных комплексов метаном также показало их эффективное взаимодействие, приводящее к образованию азота (табл. 3).
Таблица 3.
Скорости выделения азота (мол/(м2 с)) под воздействием смесей с метаном на Сu/ZSM-5 и Co/ZSM-5, рассчитанные из разных данных при 280°С
| Образец | ${{{v}}_{1}}$ × 10–12 | ${{{v}}_{2}}$ × 10–12 | ${{{v}}_{3}}$ × 10–13 | ${{{v}}_{4}}$ × 10–13 |
|---|---|---|---|---|
| Сu/ZSM-5 | 8 (410°С) | 4 | 2 | 1 |
| Co/ZSM-5 | 20 | 140 | 1.3 | 1.3 |
Как видно из данных этой таблицы, в случае системы Co/ZSM-5 наблюдается близость значений скоростей образования азота, определенных из данных различных экспериментов. Это является доказательством того, что стадия взаимодействия прочносвязанных поверхностных нитратов с метаном является скорость-определяющей стадией процесса селективного восстановления. Для системы Cu/ZSM-5, по-видимому, лимитирующей стадией является другой процесс. Также можно заметить, что для системы Cu/ZSM-5 скорость выделения азота, определенная из данных импульсных экспериментов, намного превосходит скорость стационарного катализа что, по-видимому, объясняется тем, что в промежутках между импульсами, порядка 4 мин, происходит выделение NO, который впоследствии превращается в молекулярный азот с участием органических интермедиатов на поверхности.
Превращение нитратных комплексов под воздействием пропилена было детально исследовано как для диоксида циркония, так и глин со столбиками из ZrO2, в том числе с нанесенными активными компонентами [62]. Как следует из этих данных, и для таких систем скорости превращения нитратных комплексов и каталитической реакции близки, что указывает на ключевую роль данных интермедиатов и в этом случае.
Очевидное увеличение реакционной способности нитратных и нитроорганических комплексов в ряду ZrO2–ZrPILC-Pt, Cu/ZrPILC указывает на важную роль, которую играют платина и медь в активации NO и углеводородов с образованием нитрит-нитратов, оксигенатов и трансформации C,N,O-содержащих интермедиатов в окислительно-восстановительных стадиях, что будет рассмотрено далее. Близкие константы стадий трансформации нитратных комплексов и нитроорганических соединений (табл. 4) предполагают, что каталитическая реакция может контролироваться обоими стадиями.
Таблица 4.
Характеристики трансформации нитратных комплексов под воздействием пропилена на диоксиде циркония, глине со столбиками ZrO2 (ZrPILC) и с нанесенным активным компонентом Pt + Cu (Pt,Cu/ZrPILC) [56, 62]
| Образец | T, °C | k, мин–1 | W × 10–18, молек. мин–1 |
$v$ × 10–18, молек. мин–1 |
k1, мин–1 |
|---|---|---|---|---|---|
| ZrO2 | 350 | 0.038 | 5 | 5.6 | 0.032 |
| ZrPILC | 250 | 0.035 | 2 | 1.8 | 0.040 |
| Pt,Cu/ZrPILC | 150 | 0.070 | 2.7 | 3 | 0.070 |
1.3. Маршруты трансформации нитрит-нитратных комплексов и нитроорганических соединений
Детальный анализ данных ИК-спектроскопии, полученных как в стационарных условиях проведения реакций, так и в процессе титрования адсорбированных нитрит-нитратных комплексов углеводородами в смеси с кислородом, адсорбции углеводоров из смеси с кислородом и их взаимодействия со смесью кислорода и NO, адсорбции нитроорганических соединений, в том числе с использованием изотопзамещенных молекул NO позволил установить скорости и маршруты трансформации различных поверхностных соединений и сформулировать схемы механизма реакций. В качестве типичного примера на рис. 1 приведена динамика изменения интенсивностей полос поглощения в процессе титрования нитратных комплексов для катализатора Cu-ZSM-5 [15].
Рис. 1.
Титрование нитратных комплексов на поверхности катализатора CuZSM-5 при 280°C смесью пропан + кислород (общее давление 8 Торр, О2/пропан = = 10) в статической кювете с последующей откачкой и добавлением кислорода.
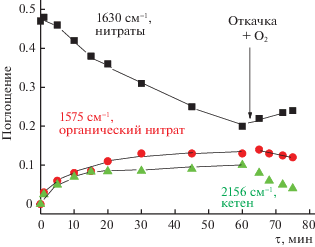
Скорость накопления органических нитратов имеет первый порядок по отношению к поверхностным нитратам с сопоставимой скоростью превращения, в то время как скорость накопление кетена имеет второй порядок. В табл. 5–7 приведены спектральные характеристики поверхностных комплексов для различных катализаторов.
Таблица 5.
Отнесение полос поглощения в спектрах после взаимодействия смеси 0.2% C3H6 + 2.5% O2 в N2 с поверхностью столбчатой глины Pt,Cu/ZrO2-PILC [56]
| Положение п.п., см–1 | Комплекс |
|---|---|
| 1672, 1419, 1370, 1240 | Координационно-связанный ацетон |
| 1392, 1381, 1465, 1335, 2979, 2938, 2870 | Изопропоксид |
| 1555, 1445 | Ацетат |
| 1625 | Н2Оадс |
| 2047, 1830 | Pt–СОадс |
Таблица 6.
Основные типы C,N,O-содержащих интермедиатов реакции селективного восстановления оксидов азота пропаном и пропиленом, обнаруженных с использованием ИК-спектроскопии
| п.п., см–1* | Соединение | Тип колебаний | Катализатор | Восстановитель |
|---|---|---|---|---|
| 2155 (2155, 2107) | кетен C=C=O | ν(C=O) | CuZSM-5 | пропан |
| 1575 (1550) | нитропропан | νas(NO2) | Cu, CoZSM-5 | пропан |
| 1380 (1361) | νs(NO2) | (Pt + Cu)/ZrPILC | пропилен | |
| 1575 | динитропропан | νsNOO | (Pt + Cu)/ZrPILC | пропилен |
| 1386 | νasNOO | |||
| 1435, 1340 | δC–H | |||
| 2730, 2887, 2945, 2985 | δC–H νC–H |
|||
| 1565 (1530) | нитропропан | νas(NO2), | ZrO2 | пропилен |
| 1380 | νs(NO2) | |||
| 1593 1328 |
нитропропан | νas(NO2) νs(NO2) |
CuO/Al2O3 | пропилен |
| 2180 (2140) | нитрил | ν(CN) | CoZSM-5 | пропан |
| 2260 (2240) | изоцианат | ν(NCO) | CoZSM-5 | пропан |
| 1662 | нитритопропан | – | CuO/Al2O3 | пропилен |
| 2237 | Cu+–NCO | ν(NCO) | CuO/Al2O3 | пропилен |
| 2156 | Cu0–CN | ν(CN) | CuO/Al2O3 | пропилен |
| 1675, 1638 | акролеин | ν(C=O), ν(C=C) |
(Pt + Cu)/ZrPILC | пропилен |
Таблица 7.
Отнесение полос поглощения в спектрах после взаимодействия смеси 0.2% NO + 0.2% C3H6 + N2 с поверхностью столбчатой глины (Pt + Cu)/ZrPILC при 150°C [56]
| п.п., см–1 | Комплекс | Отнесение |
|---|---|---|
| 1670 1420 1367 1240 |
Координационно связанный ацетон | νС=О δas–CH3 δs–CH3 νС–C |
| 2236 | Изоцианат | νN–С–O |
| 1617 1257 |
Мостиковый нитрат | |
| 1588 1248 |
Бидентатный нитрат | |
| 1575 1386 1340 1435 2770 2890 2940 2985 |
Нитроорганический комплекс (динитропропан или β-нитропропилнитрит) |
νNOO νNOO δC–H δC–H νC–H νC–H νC–H νC–H |
| 3160 | NHx | νN–H |
Было показано, что нанесение Pt и Cu на ZrO2-столбцы в составе столбчатой глины приводит лишь к количественным отличиям в адсорбционных и каталитических свойствах в реакции селективного восстановления оксидов азота пропиленом в избытке кислорода по сравнению с немодифицированными столбцами ZrO2. Как и на немодифицированной столбчатой глине, в спектрах присутствуют мостиковый и бидентатный нитратные комплексы, причем концентрация нитратных комплексов на Pt,Cu/столбчатой глине выше при прочих равных условиях. При взаимодействии с реакционной смесью 0.2 об. %C3H6 + 2.5 об. %O2/N2 на поверхности Pt,Cu/столбчатой глины присутствуют три типа комплексов: изопропоксидный комплекс, координационно-связанный ацетон и ацетат. Однако, нанесение на столбцы из ZrO2 Pt и Cu приводит к значительному понижению температуры существования изопропоксидного комплекса. Кроме того, на Pt,Cu/столбчатой глине концентрация ацетатных комплексов существенно выше, чем на немодифицированной глине. На модифицированных столбцах ZrO2 при относительно низких температурах изопропоксидный и нитратные комплексы образуют на поверхности комплекс, близкий по строению к структуре адсорбированного динитропропана, который расходуется при взаимодействии с поверхностными нитратами. В отсутствие оксидов азота и кислорода в газовой фазе и при практически полном отсутствии нитратных комплексов на поверхности преобладает реакция разложения динитропропанового комплекса, которая приводит к образованию на поверхности ацетатных комплексов и аммиака. При повышенных температурах на модифицированных столбцах ZrO2 ацетатный комплекс при взаимодействии с нитратными образует поверхностный нитрометановый комплекс, который при взаимодействии с поверхностными нитратами дает продукты реакции – молекулярный азот и СО2. Обнаруженные различия в формах активации реагентов и их термостабильности объясняют различия в активности немодифицированных и модифицированных наноразмерных столбцов ZrO2 в составе столбчатой глины в процессе селективного каталитического восстановления оксидов азота пропиленом в избытке кислорода [22, 38, 55, 56].
Для систем Cu-ZSM-5 и Co-ZSM-5 была количественно оценена реакционная способность продуктов неполного превращения нитрометана в реакции с компонентами газовой смеси реакции СКВ, такими как кислород и NO [20, 23]. На основании полученных кинетических данных сделано заключение о том что изоционаты и меламин может выступать в роли интермедиатов реакции СКВ, причем в стадии их образования из нитрометана принимают участие протоны цеолитного каркаса. Продемонстрировано участие изоцианатов как интермедиатов процесса образования и разложения меламина. Показано влияние введенных катионов на реакционную способность образующихся интермедиатов, что согласуется с предположениями, сделанными выше.
На основании полученных данных были предложены схемы механизмов реакций селективного каталитического восстановления оксидов азота углеводородами в избытке кислорода. В качестве примера на рис. 2 приведена такая схема для катализаторов на основе цеолитов ZSM-5 [15, 20].
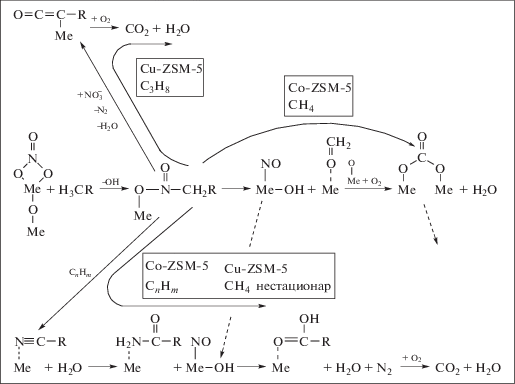
2. ПРИРОДА АКТИВНЫХ ЦЕНТРОВ
2.1. Носители
Принимая во внимание приведенные выше результаты по исследованию механизма реакции СКВ УВ, понятие активного центра в данном обзоре означает координационно-ненасыщенный катион, на котором стабилизируются реакционноспособные формы реагентов (кислород, нитрит-нитратные комплексы), и который сам способен активировать молекулу углеводорода как Льюисовский кислотный центр. Поскольку даже сами носители – диоксид циркония и γ-Al2O3 способны обеспечивать достаточно высокие уровни конверсии оксидов азота при их селективном восстановлении углеводородами в высокотемпературной области, в том числе в присутствии воды в реакционной смеси, оказывающей даже положительное влияние для оксидноциркониевой системы [33, 37, 60], представляет несомненный интерес прокомментировать природу активных центров и для этих систем.
Для обоих оксидов было показано, что активация углеводородов протекает без участия нитрит-нитратных групп, по всей видимости, на льюисовских кислотных центрах – координационно-ненасыщенных катионах алюминия и циркония [12, 24, 29]. В значительной мере именно трудность активации углеводородов на подобных центрах объясняет необходимость высоких температур для проявления заметной каталитической активности этих оксидов. Для окиси алюминия наиболее вероятными центрами являются тетракоординированные катионы алюминия, проявляющие наибольшую кислотность. Такие катионы характерны для граней (100) и (111) оксида алюминия со шпинельной структурой [55]. Кроме того, подобные низкокоординированные центры могут возникать и на других гранях вследствие наличия дефектов. Активированные углеводородные фрагменты взаимодействуют с нитрит-нитратными группами, образуя органические нитропроизводные. Высокая термическая стабильность бидентатных нитратных комплексов (температура максимума десорбции ~500°С) [22] позволяет сохранять достаточно высокие покрытия поверхности этими ключевыми интермедиатами при температурах, когда активация углеводородов протекает достаточно легко, и, следовательно, обеспечить реализацию маршрута селективного восстановления. Торможение окисления углеводородов в присутствии оксида азота в смеси с кислородом для этих систем [24, 29] позволяет полагать, что нитратные комплексы также стабилизируются на координационно-ненасыщенных катионах поверхности.
Для высокодисперсных образцов диоксида циркония концентрация и свойства активных центров поверхности – координационно-ненасыщенных катионов Zr4+ могут быть изменены как за счет введения модифицирующих низкозарядных катионов [30–34], так и за счет изменения природы фазы (моноклинная, тетрагональная или кубическая) и размеров частиц [29, 41, 42].
Введение легирующих катионов (щелочноземельных металлов – кальция, стронция и бария, а также катионов алюминия) позволило не только стабилизировать фазу тетрагонального диоксида циркония и удельную поверхность образцов, но и изменять как концентрацию координационно-ненасыщенных центров, так и их свойства [30–33, 35]. При этом в большинстве случаев оптимальное количество промотора, приводящее к высокой плотности координационно-ненасыщенных центров поверхности, не превышает нескольких атомных процентов. Для катионов алюминия при их относительно больших (более 10%) содержаниях происходит частичное блокирование поверхности частиц диоксида циркония сегрегированными оксидно-алюминиевыми кластерами [35]. Для катионов кальция и стронция увеличение их содержания приводит к реконструкции поверхностного слоя частично стабилизированного диоксида циркония в перовскитоподобную структуру, что сопровождается падением поверхностной концентрации как Бренстедовских, так и Льюисовских кислотных центров и снижением активности в реакции селективного каталитического восстановления оксидов азота пропаном (табл. 8). В то же время, для более крупных катионов бария такая реконструкция энергетически невыгодна, поэтому активность растет с увеличением содержания данного катиона (табл. 8) [30–32].
Таблица 8.
Влияние содержания допирующего катиона на активность частично стабилизированного диоксида циркония реакции селективного каталитического восстановления оксидов азота пропаном [30]
| Константа скорости |
Природа щелочноземельного катиона и его содержание (ат. %). | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ca | Sr | Ba | ||||||||
| 1.5 | 5 | 10 | 25 | 2 | 5 | 10 | 5 | 10 | 25 | |
| k × 103, 550°C | 20 | 12 | 9 | 6 | 21 | 40 | 10 | 5 | 15 | 17 |
Следует отметить, что сам процесс активации углеводородов на рассматриваемых оксидах вряд ли лимитирует процесс селективного каталитического восстановления оксидов азота, по крайней мере, при относительно малых нагрузках. Наиболее очевидно этот вывод следует из сопоставления рядов удельных активностей катализаторов в реакции селективного восстановления оксидов азота и полного окисления [60]. При близких удельных скоростях полного окисления бутана активность оксида алюминия в реакциях СКВ ниже активности оксидов циркония более чем на порядок. Не исключено, что для этих систем общая скорость процесса может лимитироваться стадией образования нитрит-нитратных комплексов, скорость которой существенно зависит от природы координационно-ненасыщенных центров поверхности и их концентрации [29, 33].
2.2. Кластерирование катионов переходных металлов и реальная структура активного компонента на носителе
Для катионов переходных металлов на носителях – оксиде алюминия и оксиде циркония [29, 33, 35, 38–40, 60], а также введенных в катионзамещенные цеолиты и в столбчатые глины [15, 23, 24, 28, 49, 50, 56, 59–61], в сравнении с соответствующими носителями, область эффективной работы в реакциях селективного восстановления оксидов углеводородами обычно смещается к низким температурам. Это явление определенно коррелирует с увеличением скорости активации углеводородов по более эффективному окислительному маршруту с образованием радикалов или продуктов селективного окисления с участием кислорода, локализованного на катионах переходных металлов [15, 19, 22, 23, 29, 33, 60]. С другой стороны, более эффективное низкотемпературное селективное восстановление оксидов азота углеводородами на нанесенных катионах переходных металлов по сравнению с самими носителями коррелирует с меньшей прочностью связи и, следовательно, большей реакционной способностью нитрит-нитратных комплексов [12, 15, 22, 33]. Поэтому несомненно, что для данных систем активный центр представляет из себя катион переходного металла, причем реакционная способность локализованных на нем реагентов зависит от характера ближайшего окружения такого катиона, определяемого свойствами носителя и степенью кластерирования катионов на данном носителе.
Как правило, для оксида алюминия, в сравнении с другими указанными выше типами носителей, катионы переходных металлов проявляют заметную активность при более высоких температурах, причем при этом достигаются более высокие степени конверсии оксидов азота. Увеличение содержания катионов меди и никеля на оксиде алюминия приводит к низкотемпературному смещению максимума активности, однако, сама величина максимальной конверсии падает [29, 33, 38, 40]. Для данных систем увеличение содержания катионов переходных металлов на поверхности приводит к росту степени их кластерирования, вплоть до образования наночастиц дисперсных оксидов. Связанное с этим увеличение скорости нецелевой реакции глубокого окисления (сжигания) углеводородов приводит к падению содержания восстановителя в слое катализатора, что и рассматривается обычно как главная причина подобного явления. Действительно, массивные оксиды переходных металлов – хорошие катализаторы реакций глубокого окисления, практически не ведут реакции селективного восстановления оксидов азота углеводородами (см. обзоры [1–11]). Однако, для катионов кобальта повышение их содержания на поверхности оксида алюминия даже в области малых концентраций [34, 60] приводит к снижению удельной (на атом кобальта) скорости реакции селективного восстановления оксидов азота и скорости окисления пропана, причем даже в большей степени для последнего (табл. 9).
Таблица 9.
Восстановление NO и окисление пропана в ходе реакции СКВ NO на Cu/Al2O3 и Co/Al2O3, состав газовой смеси: 0.1% NO, 0.13% C3H8, 1% O2 и остальное He, нагрузка 4000 ч–1. АКА рассчитана при 700 К [34]
| Катализатор | АКА × 104, молек. NO/атом Со с | АКА × 104, молек. пропана/атом Со с |
|---|---|---|
| 0.7%Co/Al2O3 | 12.1 | 3.0 |
| 3.0% Co/Al2O3 | 1.8 | 0.1 |
В то же время, для катионов меди и кобальта в структуре цеолита ZSM-5 (рис. 3) картина зависимости удельной активности от содержания введенных катионов меди или кобальта более сложная, определяемая температурой реакции. В то время как при 210°С с ростом содержания катионов наблюдается небольшое снижение активности, при 400°С наблюдается ее рост для обоих систем [15]. Поскольку при этом наблюдается снижение концентрации нитритных комплексов и рост концентрации нитратных [15], причем последние более прочно связаны (см. выше), наиболее простое объяснение заключается в том, что при более высоких температурах в реакционной среде на поверхности катализаторов доминируют нитратные комплексы, связанные с кластерами катионов переходных металлов, которые и определяют активность катализаторов в этих условиях. Соответственно снижение низкотемпературной активности коррелирует со снижением покрытия поверхности нитритными комплексами, наиболее реакционноспособными в данных условиях.
Рис. 3.
Энергия активации, удельная скорость селективного восстановления NO пропаном при 210 и 400°C в зависимости от содержания меди (а) или кобальта (б) в ZSM-5 [15].
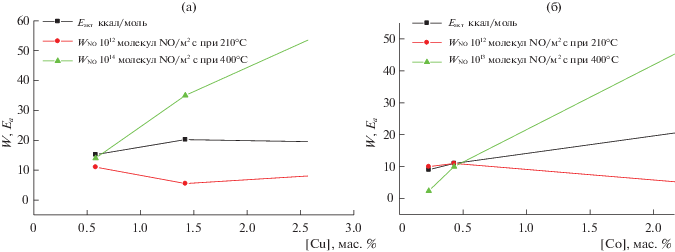
Очевидно, такие особенности связаны со спецификой структуры кластеров катионов переходных металлов на поверхности различных носителей, зависящей как от природы катиона (энергии предпочтения к тем или иным координационным окружениям), так и от структуры поверхности носителя. Проблемы кластерирования катионов переходных металлов на поверхности носителей на основе оксида алюминия и диоксида циркония были рассмотрены на примерах оксидномедной, оксидноникелевой и оксиднокобальтовой систем.
Детальное исследование состояния катионов меди на оксиде алюминия и диоксиде циркония с использованием методов ТЕМ, РФЭС, ЭСДО, ЭПР, ИКС адсорбированной молекулы-теста СО позволило выделить несколько типов центров на поверхности оксидных носителей [53, 54, 58]. Они включают в себя изолированные катионы меди (Cu2+); двумерные кластерные образования, локализованные на наиболее развитых гранях оксида алюминия типа (110); трехмерные кластеры со структурой типа NaCl, локализованные на слабо представленных гранях оксида алюминия типа (100), его неоднородностях типа поверхностных ступенек, а также на наиболее развитых гранях типа (111) кубического диоксида циркония. Концентрация изолированных катионов очень мала (не более нескольких процентов от монослоя) и быстро падает с ростом содержания активного компонента. Данные центры могут представлять собой катионы меди (Cu2+), внедренные в катионные вакансии на поверхности носителя. В соответствии с данными ЭПР [54], они имеют координацию типа плоского квадрата или искаженного октаэдра. Для катионов меди на поверхности оксида алюминия наиболее представлены плоские двумерные кластеры, которые покрывают основную часть поверхности шпинельного носителя γ-Al2O3, представленную в основном гранями типа (110). Как следует из строения данных граней, катионы Cu2+, входящие в состав таких кластеров, координированы четырьмя атомами кислорода-носителя в плоскости поверхности, одним атомом кислорода в подповерхностной позиции и одним прочносвязанным мостиковым кислородом в надповерхностной позиции. В результате такие катионы меди координационно насыщены и не способны удерживать СО даже при температуре жидкого азота. Только термовакуумная обработка или высокотемпературное восстановление в СО приводят к удалению такого кислорода с образованием координационно-ненасыщенных катионов меди Cu1+, способных удерживать СО даже при комнатной температуре. Сходная локальная структура кластеров, согласно данным ЕХАФС [15], имеет место и для Cu-ZSM-5 с высокими степенями обмена. Тем не менее, есть и существенное различие. Для медьзамещенных цеолитов весь кислород в экваториальных позициях кластеров является внерешеточным, и он может быть относительно легко удален путем восстановления водородом с образованием металлической меди, хотя это происходит труднее, чем восстановление изолированных катионов или трехмерных оксидных кластеров. Оксидномедные кластеры на поверхности оксида алюминия содержат катионы меди, внедренные в катионные вакансии носителя, и относительное содержание внерешеточного кислорода существенно меньше. В принципе, это означает большую стабильность степени окисления (1+) для последнего случая, равно как и большую среднюю прочность связи кислорода в координационной сфере меди. Даже для внерешеточного мостикового кислорода энергия связи больше в случае кластеров на оксиде алюминия на ~70 кДж/моль [15, 54, 58]. Симбатно с прочностью связи медь–кислород изменяется и термическая стабильность локализованных на таких кластерах мостиковых нитратных комплексов – при одинаковых скоростях нагрева для Cu-ZSM-5 температура максимума десорбции ~400°С, а для CuO/Al2O3 ~ 480°C [15, 16].
Наиболее интересными структурами на поверхности оксидных носителей являются трехмерные кластеры катионов меди, представляющие собой предшественников фазы оксида меди. Характерной особенностью таких структур является легкая потеря кислорода при вакуумной тренировке или под воздействием СО даже при температуре жидкого азота [53, 54, 58]. Для оксида алюминия как носителя концентрация таких кластеров невелика, и общее число входящих в них центров не превышает нескольких % монослоя. В то же время, для массивных флюоритоподобных (диоксид циркония) носителей основная часть катионов меди находится в составе таких кластеров. Легкость их восстановления в первую очередь определяется их метастабильной структурой (вероятно, типа NaCl), что позволяет образовываться связям медь–медь при удалении кислорода, облегчая, таким образом, его отрыв. Легкость восстановления таких структур коррелирует с меньшей прочностью связи с ними нитратных комплексов, десорбирующихся при температурах ~380°С [54].
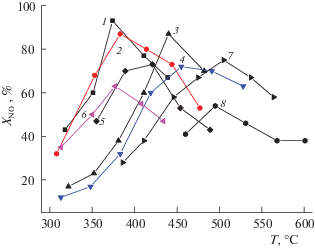
Для реакции селективного восстановления оксидов азота углеводородами как для Cu-ZSM-5 [15, 23, 28], так и для катионов меди, нанесенных на оксид алюминия или диоксид циркония [33–35, 48, 49], среднетемпературная (~350°С) каталитическая активность в реакции селективного восстановления оксидов азота пропаном возрастает с увеличением содержания активного компонента (рис. 3) до появления фазы оксида меди (содержание не более 5 мас. % меди). Для данной реакции важны как центры стабилизации нитратных комплексов, так и центры активации углеводородов. Для медьзамещеннных цеолитов обе функции играют плоские кластеры, для которых восстановительное присоединение оксида азота с образованием нитратных комплексов, сопровождаемое релаксацией структуры, ведет к возникновению координационно-ненасыщенных катионов меди (1+), способных активировать как насыщенные углеводороды типа пропана, так и ненасыщенные углеводороды [15]. Для оксидной медноалюминиевой системы возможно разделение функций между кластерами разного типа: двумерные кластеры стабилизируют нитратные комплексы, трехмерные, имеющие высокую степень координационной ненасыщенности даже в присутствии кислорода в газовой фазе, равно как и содержащие слабосвязанный кислород, преимущественно активируют углеводороды. Поскольку концентрация кластеров обоих типов растет с ростом концентрации активного компонента, наблюдается и рост удельной каталитической активности в реакции селективного восстановления оксидов азота пропаном. Меньшая прочность связи кислорода с трехмерными оксидномедными кластерами явно коррелирует с более низкими температурами эффективной работы катионов меди на поверхности диоксида циркония (температура максимальной конверсии оксидов азота порядка 300–350°С) по сравнению с оксидной медноалюминиевой системой (температура максимальной конверсии ~500–600°С, в зависимости от состава реакционной смеси (рис. 4). В этом отношении система Cu–ZrO2 близка по своей эффективности к системе Cu-ZSM-5 [33, 35]. В то же время, удельная каталитическая активность катионов меди на оксидноалюминиевом носителе как в реакции селективного каталитического восстановления оксидов азота (рис. 4), так и в реакциях полного окисления углеводородов молекулярным кислородом [15, 33, 60] значительно уступает активности катионов меди в цеолитах. Учитывая близкие энергии активации каждой реакции для катионов меди на разных носителях [60], можно полагать, что различие в эффективности в основном определяется существенно большей долей координационно-ненасыщенных катионов меди для малых кластеров в каналах цеолита и на поверхности частично стабилизированного диоксида циркония в сравнении с протяженными эпитаксиальными двумерными кластерами на поверхности оксида алюминия.
Рис. 4.
Температурные зависимости конверсии (X) NO в N2 на образцах CuO/Al2O3–ZrO2 (1–6) и CuO/Al2O3 (7, 8). Содержание меди 1 мас. % (7, 8); 2 мас. % (1–4, 6) и 4% (5). Смесь 0.1% NO + 0.1% пропана + 1% O2 в He, объемная скорость – 13000/ч (5, 8) и 4000/ч во всех других случаях. Содержание оксида алюминия в диоксиде циркония: 1% (1), 5% (2) и 10% (3–6).
Для катионов меди, фиксированных на поверхности наноразмерных столбиков диоксида циркония в столбчатых глинах [38–41, 44, 56, 59, 61], специфической особенностью является их внедрение внутрь таких наночастиц, что обусловлено рыхлой структурой последних. В результате для таких наносистем тенденция к образованию трехмерных оксидномедных кластеров выражена существенно слабее, чем для массивных флюоритоподобных систем, что отражается в существенно большей средней прочности связи кислорода и его меньшей реакционной способности по отношению к активации углеводородов. Такая специфичность структуры, а именно, большая координационная насыщенность катионов меди, отражается также в доминировании нитритных комплексов как форм адсорбции оксидов азота, легко десорбирующихся до 250°С [41, 61]. Низкое покрытие поверхности этими ключевыми интермедиатами при более высоких температурах, когда скорость активации пропана становится заметной, приводит к подавлению маршрута СКВ. В результате, для реакции селективного каталитического восстановления оксидов азота пропаном, при одном и том же содержании меди, активность в СКВ реакции катионов меди на поверхности массивного диоксида циркония [33] существенно превосходит активность катионов меди, внедренных в наночастицы диоксида циркония в столбчатых глинах [56, 61].
Для реакции селективного каталитического восстановления оксидов азота пропиленом в избытке кислорода влияние природы носителя на каталитические свойства катионов меди проявляется аналогичным образом [29]. Однако, более легкая активация пропилена по сравнению с пропаном приводит к тому, что эффект носителя в значительной мере сглажен.
Для катионов кобальта в цеолите ZSM-5 удельная каталитическая активность в реакции селективного каталитического восстановления оксидов азота метаном [15] в области температур ~400°С растет с процентом нанесения, как и для реакции восстановления пропаном (рис. 3). По данным EXAFS [15], наиболее вероятной структурой оксиднокобальтовых кластеров в цеолитах с относительно невысоким модулем Si/Al ~ 17 при содержании катионов кобальта до 2 вес. %. являются цепочки тетраэдрически координированных катионов кобальта. В этом случае доля краевых координационно-ненасыщенных катионов кобальта, на которых могут локализоваться нитрит-нитратные комплексы, достаточно велика и слабо снижается с ростом содержания кобальта. Однако, при более низких (210°С) температурах удельная каталитическая активность в реакции селективного каталитического восстановления оксидов азота метаном снижается при содержании активного компонента более 0.5 мас. %. Эта специфика объясняется тем, что с повышением содержания кобальта растет доля более прочносвязанных и менее реакционноспособных мостиковых и бидентатных нитратных комплексов, и снижается доля более реакционноспособных монодентатных нитратных комплексов [22]. При большем цеолитном модуле ~25 избыточная агрегация катионов кобальта в образцах с повышенным содержанием кобальта приводит к снижению удельной активности в реакции селективного восстановления оксидов азота пропаном при всех температурах [34].
Для катионов кобальта на оксидноалюминиевом носителе повышение концентрации кобальта от 0.7 до 3% приводило к существенному снижению удельной каталитической активности в реакции селективного каталитического восстановления оксидов азота пропаном при практически постоянной скорости окисления углеводородов в присутствии молекулярного кислорода [60]. Это позволяет исключить такую тривиальную причину снижения активности в целевой реакции как доминирование маршрута полного окисления углеводорода молекулярным кислородом. Сходные закономерности наблюдались в [67]: при повышении концентрации катионов кобальта на поверхности γ-оксида алюминия наблюдалось снижение скорости восстановления оксидов азота и падение скорости окисления углеводорода как в присутствии оксида азота, так и в присутствии только кислорода. Авторы [67] на основании дополнительных данных о координации катионов кобальта, полученных с помощью ЭСДО выдвинули предположение, что активность в селективном восстановлении оксидов азота определяется только октакоординированными в условиях контакта с окружающей средой катионами кобальта, в то время как поверхностные кластеры оксида кобальта катализируют только полное окисление углеводородов, а тетракоординированные катионы внедрены в подповерхностный слой и неактивны. Тем не менее, такое предположение явно не объясняет снижения скорости окисления углеводородов при увеличении концентрации кобальта, и, следовательно, размеров кластеров оксида кобальта. Кроме того, авторы [67] наблюдали увеличение активности и селективности расходования пропилена на восстановление оксидов азота после высокотемпературного (800°С) прокаливания образцов, когда, по их данным, происходит явное увеличение концентрации тетракоординированных катионов кобальта. Более вероятным представляется связь активности в полном окислении углеводорода молекулярным кислородом с малыми кластерами как тетра-, так и октакоординированных катионов на гранях типа (111) и (100), соответственно, которые образуются уже при самых малых концентрациях активного компонента. Это предположение согласуется с минимальной прочностью связи кислорода с такими кластерами по сравнению со всеми другими типами структур катионов кобальта на носителе. Это согласуется и с повышенной активностью катионов кобальта в цеолитах, где они имеют координацию ближнего окружения, близкую к тетраэдрической (см. выше). При объяснении падения активности в селективном восстановлении с ростом степени покрытия поверхности катионами кобальта следует принять во внимание специфику механизма реакции СКВ на оксиднокобальтовых системах, когда в присутствии оксида азота в реакционной смеси углеводород активируется непосредственно на нитрит-нитратных комплексах [15]. Поскольку изолированные катионы могут координировать монодентатные нитратные комплексы и нитритные комплексы, можно полагать, что каталитическая активность в реакции селективного каталитического восстановления оксидов азота пропаном на оксидной алюмокобальтовой системе определяется такими промежуточными прочносвязанными соединениями. В этом случае наблюдается некоторое различие ситуации для катионов кобальта на оксиде алюминия и этих же катионов, введенных в цеолит ZSM-5, когда было показано, что в близкой области температур активность в реакции восстановления оксидов азота пропаном определяется бидентатными нитратными комплексами [15, 22]. Можно полагать, что различие в природе наиболее активных форм прочносвязанных промежуточных комплексов для этих двух типов носителей определяется, в первую очередь, увеличением прочности связи кобальт-кислород для оксидной кобальт-алюминиевой системы, вследствие преимущественного координирования катионов анионами носителя. В результате бидентатные нитратные комплексы, локализованные на эпитаксиальных шпинельных оксиднокобальтовых кластерах становятся слишком прочносвязанными и малореакционноспособными в области температур ниже 500°С. В то же время, возрастание прочности связи нитритных и монодентатных нитратных комплексов делает их достаточно стабильными при рассматриваемых температурах реакции. Это предположение действительно согласуется с результатами [67], непосредственно указывающими на отсутствие разложения в условиях реакции нитрит-нитратных комплексов, стабилизированных на катионах кобальта.
Таким образом, различие природы активных центров реакции СКВ для нанесенных на оксид алюминия оксидномедных (кластеры двух типов) и оксиднокобальтовых (в основном изолированные тетракоординированные катионы кобальта) систем определяется как различием прочности связи металл–кислород, так и различием механизма реакции, в первую очередь, путей активации углеводорода. Большую роль играет также влияние матрицы на прочность связи кислорода с катионами меди и кобальта, и, следовательно, на прочность связи и реакционную способность нитрит-нитратных комплексов.
Для катионов никеля на поверхности диоксида циркония подробные исследования, проведенные в [29, 38, 40], показали, что в реакциях селективного каталитического восстановления оксидов азота пропиленом наиболее активны и селективны изолированные катионы никеля, по всей видимости, внедренные в поверхностные вакансии носителя. Вследствие достаточно высокой прочности связи кислорода с такими катионами, максимум скорости восстановления смещен в область высоких (400°С) температур. Кластерированные октакоординированные катионы-предшественники фазы оксида никеля, обладающие более высокой реакционной способностью связанного с ними кислорода, способны вести катализ при более низких температурах. Однако, максимум конверсии NО в этом случае ниже вследствие интенсивного сжигания пропилена.
2.3. Регулирование свойств катализаторов селективного восстановления оксидов азота углеводородами в избытке кислорода, содержащих катионы переходных металлов, путем введения промотирующих добавок
2.3.1. Оксидные добавки. Перспективность такого подхода была показана в самых первых работах японских исследователей [68], когда добавление катионов щелочноземельных и редкоземельных металлов в цеолиты, содержащие катионы переходных металлов, позволило существенно расширить окно рабочих температур и повысить селективность расходования углеводорода. В основном предполагалось, что это происходит за счет простого подавления маршрута полного окисления углеводородов молекулярным кислородом [68, 69]. Однако, такие эффекты могут достигаться также и за счет стабилизации интермедиатов маршрута СКВ, включая нитрит-нитратные комплексы и продукты их восстановительного превращения, содержащие атомы азота и углерода, что практически не исследовалось в известных работах.
Другой важный аспект применения промоторов связан с подавлением деалюминирования цеолита в присутствии паров воды при повышенных температурах, ведущего к потере активности за счет дестабилизации катионов переходных металлов и сегрегации их с образованием частиц оксидов. В этом случае существенную роль играет уменьшение концентрации протонов, связанных с бренстедовскими кислотными центрами, которые и ответственны за деалюминирование [69], что особенно важно для медь-замещенных цеолитов. Для кобальтзамещенных цеолитов проблема деалюминирования менее существенна, поскольку сами катионы кобальта резко увеличивают гидротермальную стабильность цеолита ZSM-5 [70]. Тем не менее, и для кобальтзамещенных цеолитов дополнительное введение катионов лантана повышало гидротермальную стабильность и активность [71].
При разработке любых реальных катализаторов на основе катионзамещенных цеолитов необходимо учитывать влияние связки, используемой либо при нанесении цеолитов на керамические или металлические блоки из суспензии, либо при экструзии массивных образцов в виде гранул или блоков. В качестве такой связки наиболее часто использовался пептизированный в кислых растворах гидроксид алюминия. Однако, использование такой связки для цеолитсодержащих катализаторов приводит к их дезактивации вследствие миграции катионов алюминия в каналы цеолита и нейтрализации бренстедовских кислотных центров или образования льюисовских центров [72]. Очевидно, этот фактор и является одной из причин существенного снижения активности блочных катализаторов на основе медьзамещенных цеолитов по сравнению с порошком цеолита. Понятно, что при подборе и введении промоторов в катализаторы селективного восстановления оксидов азота на основе катионзамещенных цеолитов необходимо учитывать также и подобные эффекты.
Исходя из определяющей роли нитрит-нитратных комплексов в маршруте реакции СКВ-НС, было выдвинуто предположение о возможности их стабилизации на поверхности при повышенных температурах за счет введения катионов щелочноземельных металлов, а также цинка, и, таким образом, обеспечить их высокую активность при больших нагрузках. Действительно, эти предположения оправдались для всех трех типов катионов переходных металлов. Сопоставление данных по ТПД оксидов азота [12, 15, 24, 25] показало, что введение катионов щелочноземельных металлов практически не меняет положение термодесорбционного пика, соответствующего разложению нитратных комплексов, однако, в несколько раз увеличивает покрытие ими поверхности. Это может указывать либо на то, что прочность связи нитратных комплексов как с катионами переходных металлов, так и с катионами промоторов близки, либо на то, что нитратные комплексы на этих двух типах центров находятся в равновесии друг с другом, по крайней мере, при повышенных температурах.
В отсутствии оксида азота в газовой фазе скорость окисления углеводорода молекулярным кислородом практически не изменилась при введении таких промотирующих добавок как стронций и цинк, что указывает на отсутствие блокирования катионов переходных металлов промотирующими катионами, равно как и на отсутствие существенного изменения прочности связи кислорода с активными центрами. Эти результаты опровергают предположение [68] о простом подавлении маршрута полного окисления как единственно возможной причине во всех случаях.
Для понимания природы влияния промоторов весьма важно то обстоятельство, что в низкотемпературной (200–300°С) области для медь- и кобальтзамещенных цеолитов при введение промоторов типа основных катионов наблюдается некоторое повышение эффективной энергии активации для реакции селективного восстановления и снижение активности [60].
Учитывая, что для немодифицированных образцов активность в этой области температур в существенной мере зависит также от нитритных комплексов, стабилизированных на изолированных катионах переходных металлов и ведущих процесс восстановления с меньшей энергией активации [15], можно полагать, что введение промотора способствует образованию бидентатных или мостиковых нитратов, стабилизированных на двух типах катионов. Действительно, для Cu-ZSM-5 и Co-ZSM-5 сопоставление данных ТПД [12, 15, 24, 25, 68, 74] показывает, что введение катиона стронция приводит к уменьшению низкотемпературного плеча на высокотемпературном пике выделения оксидов азота в экспериментах, что может указывать на снижение покрытия поверхности нитритными комплексами, связанными с изолированными катионами меди и кобальта.
Необходимо отметить, что рассмотренный механизм влияния модифицирующих катионов на активность и селективность цеолитов, содержащих катионы переходных металлов, явно не является единственным. Во-первых, сами катионы промоторов способны стабилизировать на себе моно- или бидентатные нитрит-нитратные комплексы, которые могут быть ответственны за каталитическую активность в той или иной области температур. Так, например, для образца Fe-ZSM-5, у которого катионы железа были введены в решетку в процессе гидротермального синтеза, и, судя по ТПД-спектру [12, 25], прочные формы адсорбции оксидов азота представлены только нитритными комплексами, разлагающимися с выделением NO2, введение кальция не приводит к появлению бидентатных нитратных комплексов, стабилизированных на паре разнотипных катионов. Поэтому можно полагать, что для данной системы расширение окна рабочих температур может быть связано, в первую очередь, с появлением реакционноспособных при более высоких температурах нитритных комплексов, адсорбированных на катионах кальция.
Явно другой механизм действия промотора реализуется в случае введения стронция в Cu-ZSM-5, что приводит к появлению активности данной системы в реакции селективного каталитического восстановления оксидов азота метаном [25]. Как показал анализ, проведенный в [22, 23], чрезвычайно низкая стационарная селективность расходования метана на восстановление оксидов азота для непромотированного образца Cu-ZSM-5 объясняется, в первую очередь, неспособностью катионов меди в плоскоквадратной координации удерживать в аксиальных позициях нитрометан и продукты его последующего превращения типа нитрилов и изоцианатов, обеспечивающих реализацию маршрута селективного восстановления оксидов азота. Введение основных катионов стронция, явно более способных к удерживанию анионов CN– и NCO–, чем катионы меди, в данном случае и обеспечивает их относительную стабилизацию, необходимую для протекания их последующего взаимодействия со слабосвязанными компонентами реакционной смеси или гидролиза, и, в конечном итоге, реализации маршрута спаривания атомов азота.
2.3.2. Добавка благородных металлов. Для катионов меди и кобальта на поверхности мезопористых носителей на основе частично стабилизированного диоксида циркония было детально исследовано влияние добавок серебра на активность в реакции селективного восстановления оксидов азота пропаном, пропиленом и деканом [33, 38, 40]. Существенный эффект был обнаружен только в случае использования методов приготовления, обеспечивающих сильное взаимодействие между металлическим и оксидными компонентами, что приводит к образованию смешанных кластеров. Такое взаимодействие способствует снижению прочности связи кислорода и нитрит-нитратных комплексов, что повышает их реакционную способность. В этом случае для всех трех реакций уровень активности при умеренных (250–350°С) температурах приближался к активности наиболее эффективных медь-содержащих систем, таких как Cu-ZSM-5, Cu/сульфатированный диоксид циркония, Cu/цеолит бета. Для реакций селективного восстановления оксидов азота пропиленом и деканом, когда для нанесенных оксидномедных систем поверхность в значительной степени зауглерожена, повышение окислительной способности поверхности при введении серебра препятствует такому явлению, что также является одним из факторов повышения активности.
Влияние добавок платины на активность катионов меди в реакциях восстановления оксидов азота теми же тремя восстановителями было изучено на примере систем на основе глин, пилларированных диоксидом циркония [41]. Для данного смешанного активного компонента и выбранного метода его нанесения (последовательное нанесение сначала катионов меди, затем кластеров платины) было показано, что кластеры платины преимущественно фиксируются на катионах меди, частично экранируя их. Сильное взаимодействие между металлическим и оксидным компонентом приводит к изменению состояния платины, делая ее более окисленной. В то же время, фиксация на наночастицах диоксида циркония (столбиках) катионов меди способствует ослаблению взаимодействия платины со столбиками, предотвращая ее внедрение в объем наночастиц. Взаимодействие между платиной и катионами меди влияет на способность платины активировать углеводороды, что отражается в снижении скорости окисления пропана молекулярным кислородом при его малой концентрации в смесях без оксида азота. Однако, смешанные кластеры Pt + CuOx намного менее чувствительны к негативному влиянию кислорода и NO на скорость окисления пропана в сравнении с чистыми компонентами. Увеличение степени окисления платины снижает прочность связи нитрозилов, блокирующих атомы платины, и способствует их окислению в реакционноспособные нитритные комплексы, мигрирующие на соседние катионы Cu2+ и Zr4+. Предлагаемая схема активации УВ на атомах платины с последующим взаимодействием активированных углеводородных фрагментов с нитрит-нитратными комплексами, локализованными на катионах меди на границе с кластерами платины позволяет понять причины повышенной низкотемпературной активности комбинированного активного компонента, локализованного на наночастицах диоксида циркония в столбчатых глинах.
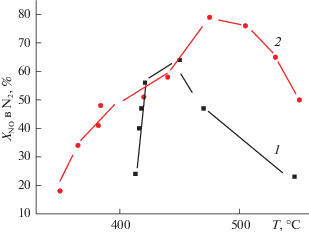
Более того, использование комбинированного активного компонента в столбчатых глинах при дополнительном модифицировании диоксидноциркониевых столбиков такими катионами как церий и железо [41, 48, 49] позволило обеспечить высокую степень конверсии оксидов азота при использовании в качестве восстановителя углеводорода с длинной цепью – декана (рис. 5). Более того, высокая эффективность сохраняется и в присутствии воды и диоксида серы в реалистических смесях.
Рис. 5.
Температурные зависимости степени превращения оксидов азота на катализаторе на основе пилларированной диоксидом циркония монтмориллонитовой глины (Fe–Zr PILC) со смешанным (Pt–Cu) активным компонентом в реакции селективного восстановления NO деканом (0.15% NO и 0.05% декана в воздухе), в том числе с добавлением в смесь 3% воды и 0.03% SO2.
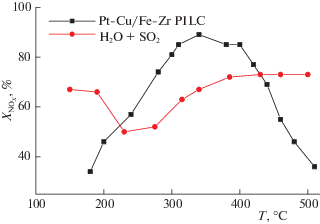
Более высокая эффективность пропилена как восстановителя отражается в более высокой конверсии оксидов азота в существенно более широком температурном интервале и при более высоких нагрузках (рис. 6). Существенным фактором является то, что комбинированный активный компонент в смесях, содержащих высокие концентрации кислорода и воды, обеспечивает высокую (90–95%) селективность образования молекулярного азота при минимальной (5–10%) селективности образования нежелательного побочного продукта – закиси азота. В то же время, в сухих смесях, содержащих небольшой избыток кислорода (содержание 1–2%), селективности по закиси азота достигала 30%.
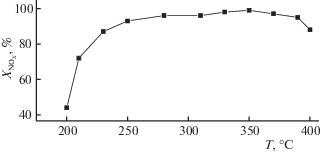
Рис. 6.
Температурная зависимость степени превращения оксидов азота на катализаторе на основе пилларированной диоксидом циркония монтмориллонитовой глине (Fe–Zr PILC) со смешанным (Pt–Cu) активным компонентом в реакции селективного восстановления NO пропиленом. Состав смеси 0.15% NO, 0.14% C3H6, 1% О2, 3% воды и 0.03% SO2, объемная скорость подачи 70 000/ч.
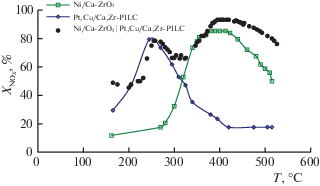
2.3.3. Комбинирование слоев катализаторов разных типов. Поскольку катализаторы на основе столбчатых глин с активным компонентом, содержащим платину, активны в низкотемпературной области, а катализаторы на основе массивного диоксида циркония наиболее активны в высокотемпературной области, была апробирована возможность комбинирования этих двух типов катализаторов в расположенных последовательно слоях [48]. При размещении в лобовом слое более высокотемпературного катализатора для реакции селективного восстановления оксидов азота пропиленом в избытке кислорода был действительно получен эффект существенного расширения температурной области эффективного удаления оксидов азота (рис. 7).
Рис. 7.
Влияние комбинирования активных компонентов двух типов в каталитическом слое на конверсию оксидов азота в процессе СКВ пропиленом в избытке кислорода [29]. Состав реакционной смеси 0.2% NO + 0.2% C3H6 + 2.5% O2 в He, объемная скорость 9000 ч–1.
2.3.4. Влияние связующего. Для промотированных образцов катион-замещенных цеолитов была подобрана связка на основе алюмосиликатов [27, 28], позволяющая сформовать их в гранулы и массивные блоки без потери высокой производительности. Для таких систем отличительным признаком является сохранение высокой конверсии оксидов азота при больших нагрузках в реальных условиях в смесях, содержащих высокие концентрации таких типичных ядов для данной реакции как вода и диоксид серы (рис. 8). Отсутствие зависимости степени конверсии оксидов азота от нагрузки для катализаторов, испытанных в виде фракции в зернистом слое, скорее всего связано с выравниванием профиля концентрации восстановителя-пропана по слою, приводящему к более эффективному восстановлению оксидов азота. Очевидно, для реализации этого явления необходима высокая скорость активации пропана, что обеспечивается достаточно высоким (2–5 мас. %) содержанием активного компонента и сильным кластерированием катионов меди. Для менее реакционноспособного метана активация на нитратных комплексах, связанных с катионами кобальта, в смесях, содержащих воду, явно протекает недостаточно быстро (рис. 9).
Рис. 8.
Влияние нагрузки (+ – 18 000 ч–1, $\blacktriangle $ – 120 000 ч–1) на конверсию оксидов азота в реакции их селективного восстановления пропаном в избытке кислорода при близких к реальному составах реакционной смеси (0.1% NO + 3%O2 + 0.2% C3H8 + 10% CO2 + 5%H2O + 2 г/м3 SO2 + N2). Модифицированный стронцием катализатор Cu-ZSM-5 сформован в виде блоков со связующим и испытан в виде фракции 0.5–1 мм.
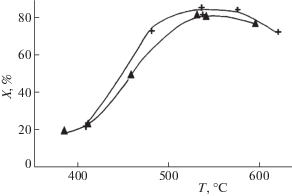
Рис. 9.
Влияние палладия на эффективность работы блочных катализаторов на основе кобальт-замещенного цеолита ZSM-5. Испытание в виде фракции, состав реакционной смеси 0.02% NO, 0.4% CH4, 16% O2, 2% H2 в He, объемная скорость 12 000/ч. Состав активного компонента: 1 – Co-ZSM-5, 2 – Co-ZSM-5 + Pd.
Для повышения активности в реакции селективного восстановления оксидов азота метаном в низкотемпературной области был использован прием введения небольших добавок палладия, способствующего эффективной активации метана, что действительно привело к расширению температурной области эффективной работы катализатора (рис. 9).
Цеолитсодержащие компоненты на основе системы Cu–Sr/ZSM-5 были нанесены на тонкостенные блоки из окисленной и термостабилизированной алюминиевой фольги и дополнительно промотированы палладием [74]. Испытания таких блоков в дизельном нейтрализаторе городского автобуса “Икарус” вместе с блочными катализаторами, предназначенными для удаления жидких частиц дыма, СО и углеводородов показали, что в течение по крайней мере 4 месяцев достигался уровень удаления оксидов азота не менее 50%.
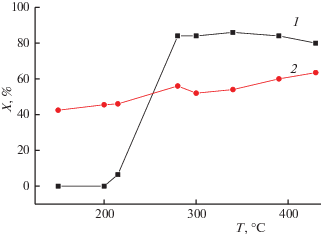
Для систем на основе столбчатых глин и каркасных фосфатов циркония, содержащих катионы меди и промотированные платиной, была разработана процедура их нанесения на кордиеритовые тонкостенные блоки с плотностью 400 ячеек на квадратный дюйм. Для этого использовались суспензии активного компонента со связующим на основе гидроксида алюминия или оксинитрата циркония [49, 51]. Как следует из результатов, представленных на рис. 10 и 11, в реакции восстановления оксидов азота деканом в избытке кислорода, даже в смесях с большим содержанием воды, на микроблоках, испытанных в проточном реакторе, достигаются конверсии оксидов азота и декана, сопоставимые с лучшими результатами на нанесенных платиновых катализаторах, разработанных корпорацией Мазда Мотор [75, 76]. В указанных катализаторах Мазда Мотор используется комбинация Pt, Rh и Ir, введенных в цеолит H-ZSM-5, который наносится на кордиеритовые блоки. При введении в качестве добавки диоксида церия конверсия оксидов азота достигает 70%. Однако, старение катализатора в жестких условиях приводило к снижению показателей, по всей видимости, вследствие термодинамической нестабильности структуры цеолита. В то же время, для разработанных нами катализаторов такие явления не были отмечены.
Рис. 10.
Температурные зависимости конверсии декана (1) и оксидов азота (2) на микроблочном катализаторе (Pt–Cu/Zr PILC/кордиерит, связующее на основе алюмосиликатов) в реакции селективного восстановления оксидов азота деканом. Состав смеси 0.05% декана, 0.1% NO, 10% H2O и 10% O2 в гелии, объемная скорость 25 000/ч.
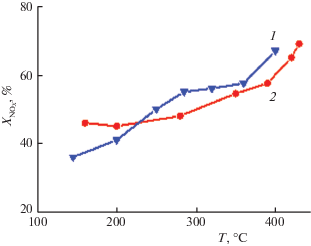
Рис. 11.
Температурные зависимости конверсии декана (1) и оксидов азота (2) на микроблочном катализаторе Pt + Cu-цирконий фосфат/кордиерит (связка на основе диоксида циркония) в реакции селективного восстановления оксидов азота деканом. Состав смеси: 0.05% декана, 0.1% NO, 10% H2O и 10% O2 в гелии. Объемная скорость 30 000/ч.
ЗАКЛЮЧЕНИЕ
Установление основных характеристик механизма реакции селективного каталитического восстановления оксидов азота углеводородами в избытке кислорода позволило подойти к решению проблемы дизайна активных компонентов катализаторов данных процессов. Основными требованиями к активному компоненту являются сочетание двух функций: (i) обеспечение достаточной прочности связи ключевых интермедиатов нитрит-нитратных комплексов с активными центрами поверхности (следовательно, достаточно высоких покрытий в условиях реального катализа), (ii) высокой скорости активации углеводородов на таких центрах и эффективного взаимодействия активированных фрагментов с нитрит-нитратными комплексами. С точки зрения дизайна, регулирование свойств центров достигается путем изменения их химической природы (тип катиона переходного металла на поверхности носителя), ближайшего координационного окружения (определяется свойствами носителя или оксидной матрицы, а также степенью кластерирования катионов на носителе). Достаточно эффективным оказываются подходы, когда функции стабилизации нитрит-нитратных комплексов и активации углеводорода оказываются разделены между оксидными и металлическими составляющими наноструктурированного активного компонента. Применение этих критериев к активным компонентам на основе катионов меди, кобальта, никеля и железа, фиксированных на таких носителях как высококремнистые цеолиты, частично стабилизированный диоксид циркония, пилларированные диоксидом циркония природные глины, в том числе промотированных серебром или платиной и каркасные фосфаты циркония позволило создать системы, обладающие высокой активностью, селективностью и стабильностью в целевых реакциях при составах отходящих газов, близких к реальным. Для их практического использования разработаны методы нанесение этих активных компонентов на блочные носители сотовой структуры или формовка их в виде блоков с использованием различных связующих. Данные катализаторы защищены патентами РФ.
Эта работа частично поддерживалась Минпромэнерго СССР, РАО ЕЭС, РФФИ, ИНТАС и Интеграционным проектом Президиума РАН 8.17. Авторы обзора искренне благодарны всем сотрудникам, принявшим участие в выполнении экспериментальных исследований и написании оригинальных статей и патентов.
Список литературы
Iwamoto M., Yahiro H., Shundo S. et al. // Shokubai (Catalyst). 1990. V. 32. P. 430.
Iwamoto M. Proc. Meeting of Catalyt. Technol. for Removal of Nitrogen Monoxide, Tokyo, 1990. P. 1.
Held W., Koenig A. Ger. Offen. DE 3 642 018 (1987).
Held. W., Koenig A., Richter T., Puppe L. SAE Paper 900496 (1990).
Iwamoto M., Mizuno N., Yahiro H. // New Frontiers in Catalysis (Proc. 10th Int. Congr. Catalysis), Akademiai Kiado. Budapest, 1993. part. B. P. 1285.
Iwamoto M. // Zeolites and Related Microporous Materials: State of the Art 1994. (Proc. 10th Int. Zeolite Conf.), J. Weitkamp, H. G. Karge, H. Pfeifer, and H. Holderich, Eds. P. 1395.
Iwamoto M. // Catal. Today. 1996. V. 29. № 1–4. P. 29.
Amiridis M.D., Zhang T., Farrauto R.J. // Appl. Catal. B: Environ. 1996. V. 10. P. 203.
Centi G., Perathoner S. // Appl. Catal. A. 1995. V. 132. P. 179.
Walker A.P. // Catal. Today. 1995. V. 26. P. 107.
Xu J., Wang H., Guo F. et al. // RSC Adv. 2019. V. 9. P. 824.
Sadykov V.A., Baron S.L., Matyshak V.A. et al. // Catal. Lett. 1996. V. 37. P. 157.
Matyshak V.A., Baron S.L., Ukharskii A.A. et al. // Kinetics and Catalysis. 1996. V. 37. P. 549.
Sadykov V.A., Paukshtis E.A., Beloshapkin S.A. et al. // Proc. 8th Int. Symp. on Heterog. Catalysis. Varna. 1996. V. 1. P. 347.
Sadykov V.A., Beloshapkin S.A., Paukshtis E.A. et al. // Pol. J. Environ. Stud. 1997. V. 1. P. 21.
Sadykov V.A., Beloshapkin S.A., Paukshtis E.A. et al. // React. Kinet. Catal. Lett. 1998. V. 64. P. 185.
Sadykov V.A., Rozovskii A.Ya., Lunin V.V. et al. // Ibid. 1999. V. 66. P. 337.
Beloshapkin S.A., Matyshak V.A., Paukshtis E.A. et al. // Ibid. 1999. V. 66. P. 297.
Matyshak V.A., Ukharskii A.A., Ilyichev A.N. et al. // Kinetics and Catalysis. 1999. V. 40. P. 116.
Beloshapkin S.A., Paukshtis E.A., Sadykov. V.A. // J. Molec. Catal. A: Chem. 2000. V. 158. P. 355.
Matyshak V.A., Il’ichev A.N., Ukharsky A.A., Korchak V.N. // J. Catal. 1997. V. 171. P. 245.
Садыков В.А., Лунин В.В., Матышак В.А. и др. // Кинетика и катализ 2003. Т. 44. С. 412.
Белошапкин С.А. Дисс. … канд. наук. Институт катализа СО РАН им. Г.К. Борескова, Новосибирск, 2000.
Sadykov V.A., Rozovskii A.Ya., Lunin V.V., Matyshak V.A. U.S.-Russia Workshop on Environmental Catalysis. Wilmington, Delaware, 1994. 20 p.
Sadykov V.A., Alikina G.M., Bunina R.V. et al. // World Congress on Envir. Catal., Pisa, Italy, 1995; G. Centy, C. Cristiani, P. Forzatti, S. Perathoner, Eds., Societa Chimica Italiana, Roma, Italy. P. 315.
Sadykov V.A., Alikina G.M., Baron S.L. et al. // Abstr. 11th Intern. Congr. Catal., Baltimore, USA. 1996. Po-202.
Romannikov V.N., Alikina G.M., Sadykov V.A. et al. Catalyst for the Process of the Nitrogen Oxides Abatement in the Exhausts and Method of its Preparation. Patent RU No 2072897 (1994).
Sadykov V.A., Bunina R.V., Alikina G.M. et al. Method of Purification of the Exhausts from the Nitrogen Oxides. Patent RU No 2088316 (1995).
Конин Г.А. Дисс. … канд. наук. Институт химической физики им. Н.Н. Семенова РАН, Москва, 2001.
Sadykov V.A., Ivanova A.S., Ivanov V.P. et al. // Mater. Res. Soc. Symp. Ser., 1997. V. 454 (Advanced Catalytic Materials), P. 199.
Ivanova A.S., Alikina G.M., Solovyova L.P. et al. // React. Kinet. Catal. Lett. 1996. V. 59. № 1. P. 125.
Kharlanov A.N., Zubareva N.A., Lunina E.V. et al. // Vestnik MGU. Ser. 2. Khimia. 1998. V. 39. № 1. P. 29.
Sadykov V.A., Bunina R.V., Alikina G.M. et al. // J. Catal. 2001. V. 200. P. 131.
Метелкина О.В., Лунин В.В., Садыков В.А. и др. // Нефтехимия. 2000. Т. 40. № 2. С. 108.
Metelkina O.V., Lunin V.V., Sadykov V.A. et al. // Catal. Lett. 2002. V. 78. P. 111.
Konin G.A., I’ichev A.N., Matyshak V.A. et al. // Europacat-V. September. 2001. Limerick. Ireland. Abstracts Book 2. 21-O-10.
Ivanova A.S., Alikina G.M., Sadykov V.A. et al. Catalyst of the Reduction of Nitrogen Oxides by Hydrocarbons and Method of its Preparation. Patent RU No 2043146 (1992).
Konin G.A., Il’ichev A.N., Matyshak V.A. et al. // Topics in Catalysis. 2001. V. 17. № 1–4. P. 193.
Sadykov V. A., Bunina R.V., Alikina G.M. et al. // Mat. Res. Soc. Symp. Proc., 2000. V. 581. P. 435.
Konin G.A., Il’ichev A.N., Matyshak V.A. et al. Proc. CAPOC-5. Brussels. 2000. V. 2. P. 97.
Sadykov V.A., Kuznetsova T.G., Doronin V.P. Proc. CAPOC-5 Chemistry for Sustainable Development 2003. V. 11. P. 249.
Sadykov V.A., Kuznetsova T.G., Doronin V.P. et al. // Mat. Res. Soc. Symp. Proc. 2002. V. 703. P. 529.
Fenelonov V.B., Derevyankin A.Yu., Sadykov V.A. // Microporous Mesoporous Materials. 2001. V. 47. P. 359.
Sadykov V.A., Kuznetsova T.G., Doronin V.P. et al. // Proc. X APAM Topical Seminar and III Conference “Materials of Siberia. Nanoscience and Technology”.June 2-6.2003. Novosibirsk. Russia. P. 208.
Sadykov V.A., Kuznetsova T.G., Alikina G.M. et al. // Europacat-V, September, 2001. Limerick. Ireland.Abstracts Book 3. 7-O-03.
Kuznetsova T.G., Sadykov V.A., Paukshtis E.A. et al. // VI Conf. “Mechanisms of Catalytic Reactions”. October 1–5. 2002. Moscow. Russia. V. 2. P. 197.
Sadykov V.A., Doronin V.P., Sorokina T.P. et al. // Abstracts, Russian – Dutch Workshop “Catalysis for Sustainable Development”. June 22–25. 2002. Novosibirsk, Russia. P. 235.
Кузнецова Т.Г., Садыков В.А., Сорокина Т.П. и др. Катализатор, носитель катализатора, способ их приготовления (варианты) и способ очистки отходящих газов от оксидов азота (варианты). Патент РФ № 2194573 от 17.09.01.
Кузнецова Т.Г., Садыков В.А., Сорокина Т.П. и др. Катализатор, носитель катализатора, способ их приготовления (варианты) и способ очистки отходящих газов от оксидов азота (варианты). Патент РФ № 2199389 от 17.09.01.
Sadykov V.A., Pavlova S.N., Chaikina M.V. et al. // Chemistry for Sustainable Development. 2002. V. 10. P. 227.
Pavlova S.N., Sadykov V.A., Zabolotnaya G.V. et al. // Abstr. 3rd Intern. Conf. Inorganic Materials. Konstanz, Germany. 7–10 September 2002. P. 179.
Kriventsov V.V., Kochubey D.I., Sadykov V.A. et al. // Nuclear Instruments and Methods in Physics Research 2001. V. 470. № 1–2. P. 336.
Sadykov V.A., Bunina R.V., Alikina G.M. et al. // J. Catal. 2001. V. 200. P. 117.
Mezentseva N.V., Bedilo A.F., Volodin A.M. et al. // Zh. Phys. Khim. 2006. V. 80. № 7. P. 1239.
Matyshak V.A., Tretyakov V.F., Chernyshev K.A. et al. // Kinetics and Catalysis 2006. V. 47. № 5. P. 747.
Sadykov V., Kuznetsova T., Doronin V. et al. // Catal. Today 2006. V. 114. P. 13.
Sadykov V.A., Kuznetsova T.G., Bunina R.V. et al. // Chemistry in Sustainable Development 2005. V. 13. P. 713.
Tikhov S.F., Sadykov V.A., Kryukova G.N. et al. // J. Catalysis. 1992. V. 134. P. 506.
Sadykov V.A., Kuznetsova T.G., Bunina R.V. et al. // Mater. Res. Soc. Symp. Proc. 2005. V. 876E.P. R8.21.1-6.
Sadykov V.A., Lunin V.V., Rozovskii A.Ya. et al. // Green Chemistry in Russia (V. Lunin, P. Tundo and E. Lokteva, Eds.), Green Chemistry Series no 12.INCA. Italy. 2005. P. 45.
Sadykov V.A., Kuznetsova T.G., Doronin V.P. et al. // Topics in Catalysis 2005. V. 32. P. 29.
Matyshak V.A., Sadykov V.A., Chernyshov K.A., Ross J. // Catal. Today. 2009. V. 145. P. 152. https://doi.org/10.1016/j.cattod.2008.04.018
Matyshak V.A., Il’ichevA.N., Sadykov V.A. et al. // Kinetics and Catalysis. 2015. V. 56. № 2. P. 226.
Matyshak V.A. // Current Catalysis. 2017. V. 6. № 2. P. 90.
Матышак В.А., Сильченкова О.Н. // Кинетика и катализ. 2019. Т. 60. № 5. C. 578.
Knozinger H., Ratnasamy P. // Catal. Rev. Sci. Eng. 1978. V. 17. P. 31.
Yan J.Y., Kung M.C., Sachtler W.M.H., Kung H.H. // J. Catal. 1997. V. 172. № 1. P. 178.
Teraoka Y., Ogawa H., Furukawa H., Kagawa S. // Catal. Lett. 1992. V. 12. P. 361.
Grinsted R.A., Jen H.-W., Montreuil C.N. et al. // Zeolites. 1993. V. 13. № 8. P. 602.
Armor J.N., Farris Th.S. // Appl. Catal. B: Environ. 1994. V. 4. № 1. P. L11- L17.
Budi P., Howe R.F. // Catal. Today. 1997. V. 38. № 2. P. 175.
Martin A., Berndt H., Lohse U., Wolf U. // J. Chem. Soc. Farad. Trans. 1993. V. 89. № 8. P. 1277.
Stakheev A.Yu., Lee C.W., Park S.J., Chong P.J. // Appl. Catal. B: Environ. 1996. V. 9. P. 65.
Tikhov S.F., Chernykh G.V., Sadykov V.A. et al. // Catal. Today. 1999. V. 53. P. 639.
Takami A., Takemoto T., Iwakuni H. et al. // Ibid. 1997. V. 35. P. 75.
Takami A., Takemoto T., Iwakuni H. et al. // SAE Trans. 1995. Section 4 № 950 746. P. 521.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии