

Журнал физической химии, 2021, T. 95, № 3, стр. 405-413
Влияние природы носителя на активность катализаторов на основе 1% Cs/Me0.1Co2.9O4 (Me = Ni, Mg) в реакции низкотемпературного разложения N2O
Л. А. Исупова a, *, Ю. А. Иванова a
a Российская академия наук, Сибирское отделение, Институт катализа им. Г.К. Борескова
Новосибирск, Россия
* E-mail: isupova@catalysis.ru
Поступила в редакцию 20.08.2020
После доработки 20.08.2020
Принята к публикации 24.08.2020
Аннотация
Методами рентгеновского анализа, адсорбции БЭТ, О2-ТПД изучено влияние природы носителя (α-Al2O3, Mg2Al4Si5O18, CeO2, TiO2) на свойства нанесенных катализаторов на основе Ni,Mg-замещенной кобальтовой шпинели. Исследована их каталитическая активность в реакции разложения закиси азота при температурах 190–400°С. Показано, что наиболее активным и устойчивым к каталитическим ядам (O2, H2O) является катализатор на основе магний-замещенной кобальтовой шпинели, нанесенной на корундовый носитель – 1%Cs/Mg0.1Co2.9O4/α-Al2O3.
Защита окружающей среды от парникового газа N2O, в 310 раз более опасного чем СО2, концентрирует внимание исследователей на способах его удаления из газовых выбросов в атмосферу. Один из основных поставщиков закиси азота в атмосферу – производство азотной кислоты, в котором закись азота образуется в качестве побочного продукта на стадии окисления аммиака [1, 2]. С учетом технологической схемы производства азотной кислоты рассматриваются три возможных способа реализации процесса удаления закиси азота, различающиеся по условиям [1, 2]: 1) высокотемпературное разложение путем размещения слоя катализатора сразу после катализаторных платиноидных сеток, по аналогии с разработанной ранее в России двухступенчатой системой процесса окисления аммиака с использованием блочного высокотемпературного катализатора [3–17]; 2) удаление закиси азота и оксидов азота в реакторе селективного каталитического восстановления (CКВ) оксидов азота [1, 2, 11–15] и 3) низкотемпературное разложение закиси азота в отходящих газах [16–18]. Возможность организации комплексной низкотемпературной каталитической очистки хвостовых газов от NOx и N2O в реакторе СКВ, когда в первом слое происходит каталитическое восстановление NOx аммиаком, а во втором слое – каталитическое разложение N2O, на сегодняшний день считается технологическим трендом. Например, компания Uhde EnviNOX® для комплексной очистки предложила двухступенчатую систему, основанную на использовании цеолитных катализаторов [11–15, 19–21]. Система работает при температуре выше 400°С и чувствительна к парам воды [22].
Основные проблемы низкотемпературного удаления закиси азота в России – это более низкая температура газов после процесса СКВ (до 300°С), очень низкие концентрации N2O (1000–3000 ppm) и наличие ингибирующих реагентов (O2, H2O, NOх, NH4), что ставит задачу разработки более низкотемпературного катализатора разложения закиси азота, который может использоваться в комбинированной схеме очистки отходящих газов после стадии селективной очистки от NOx, согласно современным тенденциям [1, 19–21]. Самыми перспективными низкотемпературными оксидными катализаторами для удаления N2O на сегодняшний день являются кобальтовые шпинели [23–27]. Увеличение активности кобальтовой шпинели достигается путем ее модифицирования такими элементами как Mg, Ni, Mn, Cu, Cr [28] и легированием щелочными катионами (Na, K, Cs) [29]. Предполагается, что это приводит к образованию вакансий, на которых формируются слабосвязанные формы кислорода, что способствует облегчению лимитирующей стадии процесса – стадии десорбции кислорода, образующегося в ходе реакции разложения закиси азота, согласно существующим представлениям о механизме данной реакции. Разложение закиси азота может протекать по двум основным механизмам: по механизму Ленгмюра–Хиншельвуда (Л–Х) в области низких температур, а в области высоких температур – по механизму Или–Ридела (И–Р) [30, 31]:
диссоциативная адсорбция N2O
(1)
${{{\text{N}}}_{{\text{2}}}}{\text{O}} + {{{\text{S}}}_{{{\text{surf}}}}} \to {{{\text{N}}}_{2}} + {\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}},$образование и десорбция О2 по механизму Л–Х
(2)
${\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}} + {\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}} \leftrightarrow 2({\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}}) \leftrightarrow 2{\text{S}} + {{{\text{O}}}_{2}},$десорбция О2 по механизму И–Р
(3)
${\text{S}} \ldots {{{\text{O}}}_{{{\text{surf}}}}} + {{{\text{N}}}_{{\text{2}}}}{\text{O}} \to {\text{S}} + {{{\text{O}}}_{2}} + {{{\text{N}}}_{{\text{2}}}},$Использование нанесенных катализаторов позволяет снизить количество активного компонента в катализаторе и его стоимость, при этом увеличить механическую прочность. Носитель может быть представлен в различных геометрических формах: блоки, кольца, гранулы или в виде структурированного материала, что технологически немаловажно. В качестве носителей обычно используются такие оксидные материалы как: TiO2, Al2O3, SiO2, ZrO2, CeO2, цеолиты и другие [25, 32–35].
Выбор носителя зависит от требований, предъявляемых катализатору. Низкотемпературное разложение закиси азота не требует высокой термической устойчивости носителя, но он должен обеспечивать высокую дисперсность активного компонента и не вступать с ним в химическое взаимодействие. Природа носителя в значительной мере влияет на активность. Так, в [36] представлен ряд активности в реакции разложения N2O для шпинели Co3O4, нанесенной на оксиды: MgO > ZnO > MnxOy > Al2O3 > CeO2. Авторы объясняют высокую активность катализатора Co3O4/MgO электронными взаимодействиями между носителем и активным компонентом, способствующим десорбции поверхностного кислорода по механизму Ленгмюра–Хиншельвуда (2).
В выполненных нами работах по исследованию влияния модифицирования и промотирования на активность кобальтовой шпинели были определены наиболее перспективные системы на основе кобальтовой шпинели, модифицированной катионами магния или никеля и промотированной 1% цезия – 1%Cs/Me0.1Co2.9O4 (Me = Ni, Mg) [37, 38].
Цель данной работы – исследование влияния носителей разной природы на активность двух нанесенных наиболее активных оксидных систем 1%Cs/Me0.1Co2.9O4 (Me = Ni, Mg) в реакции низкотемпературного разложения закиси азота. В качестве носителей были выбраны оксиды: CeO2, TiO2, α-Al2O3 (корунд) и Mg2Al4Si5O18 (кордиерит).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Нанесение активного компонента 1%Cs/Me0.1 Co2.9O4 (Me = Ni, Mg) на носители CeO2, TiO2, α-Al2O3 (корунд), Mg2Al4Si5O18 (кордиерит) проводили суспензионным методом осаждения. В качестве носителей использовали CeO2, полученный прокаливанием при 800°С в течение 4 ч Се(NO3)3 ⋅ 6H2O (“ч.д.а.”, производитель “Союзхимпромом”); TiO2 (“ч”, производитель “Вектон”); α-Al2O3 (корунд), полученный нами по методике [39]; и Mg2Al4Si5O18 (кордиерит), полученный нами по методике [40].
Количество активного компонента относительно носителя составляло 50 мас. %. В совместный раствор солей Co(NO3)2 ⋅ 6H2O (“ч”, “Реахим”) и Mg(NO3)2 ⋅ 6H2O (“ч.д.а.”, “Реахим”) либо Ni(NO3)2 ⋅ 6H2O (“ч.д.а.”, “Реахим”) объемом 100 мл помещали мелкодисперсный носитель. В полученную суспензию при комнатной температуре и постоянном перемешивании добавляли в течение 1 ч (~250 мл) 0.8M раствор (NH4)2CO3 (“х.ч.”, “Реахим”) доводя до pH 8–8.5. После осаждения солей карбонатов в смесь добавляли CsNO3, лимонную кислоту и этиленгликоль (по методике “Pechini”). Полученную суспензию далее сушили при температуре 120°С в течение 24 ч. Сухой осадок прокаливали в течение 2 ч при 500°С. Полученные нанесенные катализаторы 1%Cs/Me0.1Co2.9O4 (Me = Ni, Mg)/носитель обозначены как Cs/Ni-Co/носитель и Cs/Mg-Co/носитель.
Фазовый состав образцов определяли методом рентгенофазового анализа (РФА) на дифрактометре Bruker D8 (Германия) с использованием излучения CuKα (λ = 1.5418 Å). Образцы сканировали по точкам с интервалом 0.05° в диапазоне 2θ от 10 до 70°. Расчет структурных параметров проводили с помощью программы Powder Cell 2.4. Методом БЭТ по термодесорбции аргона определяли удельную поверхность (Sуд, м2/г) образцов.
Исследование образцов методом термопрограммируемой десорбции кислорода (O2-ТПД) выполняли в проточной установке, анализ смеси на выходе из реактора проводили с помощью масс-спектрометра QMS 100 SRS RGA. Предварительно образцы обрабатывали в смеси 20% O2 в He в течение 60 мин при 450°С, после чего охлаждали до комнатной температуры. Навеска образцов составляла 400 мг, скорость подачи Не – 3.6 л/ч, скорость нагрева – 30 К/мин до 450°С. Количество десорбируемых атомов кислорода в монослоях рассчитывали в предположении, что в монослое содержится 1 × 1019 атомов O/м2.
Каталитическую активность в реакции разложения закиси азота определяли для фракции (0.5–0.25 мм) катализаторов в проточном реакторе при температурах 190–400°С, объемной нагрузке 9000 ч–1 и составе реакционной смеси 0.15% N2O в He. Длительные испытания на устойчивость к ингибиторам проводили при температуре 300°С добавлением в исходную газовую смесь либо 3% O2, либо 2.5–3% H2O. Определение концентрации закиси азота на входе и выходе из реактора проводили в режиме on-line при температуре 120°С с помощью ИК-фурье-спектрометра “ФТ-801”. Степень конверсии рассчитывали по формуле:
Температура, при которой конверсия N2O достигала 50% (обозначена как Т50 – температура полупревращения закиси азота), служила мерой активности катализаторов.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Результаты выполненных исследований приготовленных катализаторов и образцов сравнения представлены на рис. 1–4 и в табл. 1. На рентгенограммах (рис. 1) всех приготовленных образцов присутствуют интенсивные рефлексы, соответствующие пространственной группе Fd-3M, которая типична для Co3O4, и рефлексы соответствующего носителя: α-Al2O3 (R-3CH) корунд, TiO2 (P42/MNM) рутил, CeO2 (FM-3M) флюорит и Mg2Al4Si5O18 (CCCM) кордиерит.
Рис. 1.
Рентгенограммы нанесенных катализаторов: а – Cs/Mg–Co/носитель, б – Cs/Ni–Co/носитель. Носители: CeO2, TiO2, α-Al2O3, Mg2Al4Si5O18 обозначены на рисунках.
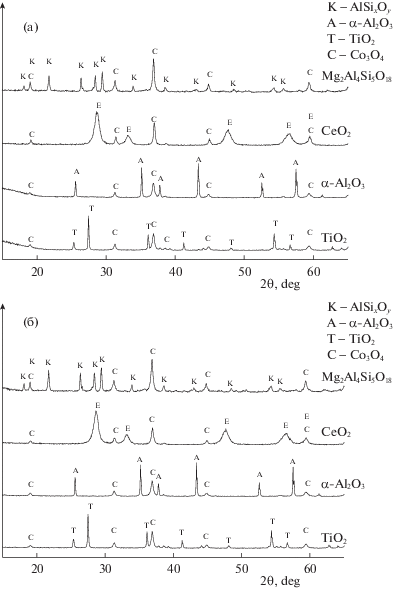
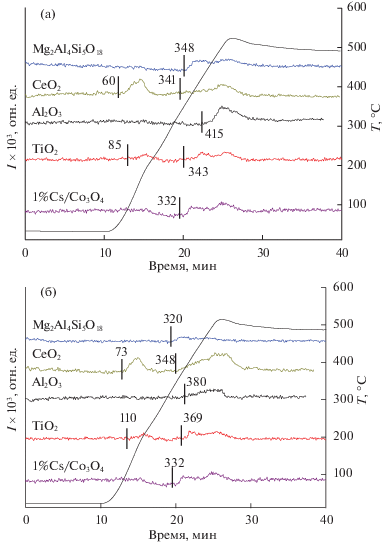
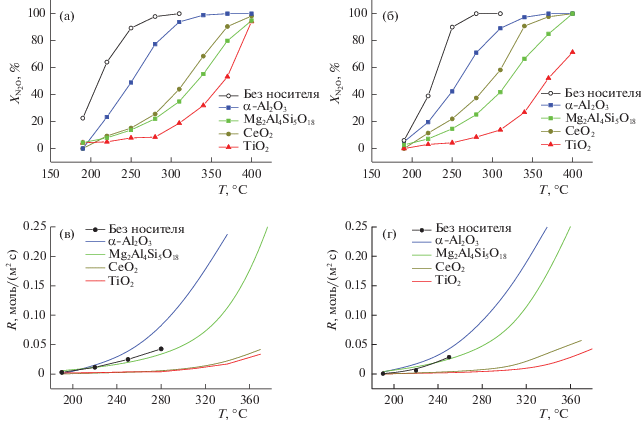
Рис. 4.
Конверсия N2O (%) при 300°С при длительных испытаниях нанесенных катализаторов в реакционных смесях, содержащих ингибиторы: а – 1500 ppm N2O, 3.6% O2 в He; б – 1500 ppm N2O, 3% Н2O в He.
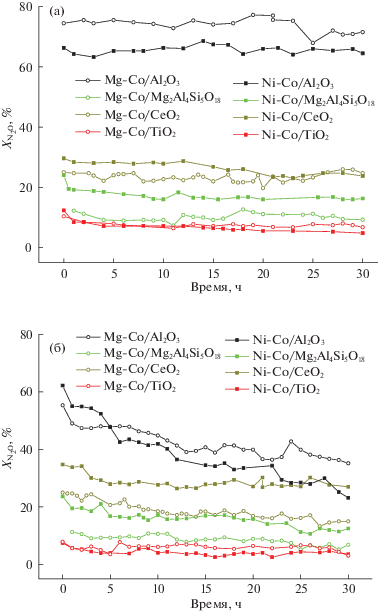
Таблица 1.
Текстурные и структурные характеристики приготовленных образцов, количество десорбирующегося кислорода (по данным ТПД) и температура достижения 50% степени конверсии закиси азота
| № | Образцы | SБЭТ, м2/г |
Т50%,оС | O2-ТПД, моль O2 × 105 (монослой) | ОКР, Å | Параметры ячейки, Å | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 пик 70–250°C |
2 пик 300–500°C |
N | Co3O4 | Носи-тель | Co3O4 (a = b = = c) |
Носитель (a) (b) (с) |
||||
| 1 | Cs/Ni–Co | 32 | 226 | 48.9 (4.6) | 0 | 4.6 | 260 | – | 8.083 | – |
| 2 | Cs/Ni–Сo/Mg2Al4Si5O18 | 5 | 320 | 0 | 1.57 (0.8) | 0.8 | 270 | 400 | 8.081 | 16.998 9.766 9.323 |
| 3 | Cs/Ni–Co/CeO2 | 53 | 298 | 2.45 (0.1) | 14.5 (0.8) | 0.9 | 220 | 100 | 8.082 | 5.411 |
| 4 | Cs/Ni–Co/α-Al2O3 | 11 | 258 | 0 | 2.71 (0.7) | 0.7 | 245 | 640 | 8.077 | 4.758 12.990 |
| 5 | Cs/Ni–Co/TiO2 | 17 | 367 | 0.72 (0.13) | 2. 85 (0.5) | 0.7 | 250 | 470 | 8.079 | 4.593 2.959 |
| 6 | Cs/Mg–Co | 36 | 210 | 68.2 (5.7) | 0 | 5.7 | 210 | – | 8.084 | – |
| 7 | Cs/Mg–Co/Mg2Al4Si5O18 | 6 | 332 | 0 | 2.85 (1.7) | 1.7 | 340 | 445 | 8.083 | 17.005 9.780 9.330 |
| 8 | Cs/Mg–Co/CeO2 | 45 | 317 | 2.99 (0.20) | 13.9 (0.9) | 1.1 | 300 | 90 | 8.086 | 5.411 |
| 9 | Cs/Mg–Co/α-Al2O3 | 15 | 252 | 0 | 8.55 (1.7) | 1.7 | 220 | 515 | 8.080 | 4.758 12.990 |
| 10 | Cs/Mg–Co/TiO2 | 18 | 366 | 1.19 (0.20) | 2.85 (0.5) | 0.7 | 240 | 530 | 8.082 | 4.593 2.959 |
| 11 | 1%Cs/Co3O4 | 28 | 275 | 0 | 2.15 (0.2) | 0.2 | 380 | – | 8.083 | – |
Рассчитанные параметры кристаллической ячейки кобальтовой шпинели в нанесенных катализаторах и величины областей когерентного рассеяния (ОКР), приведенные в табл. 1, свидетельствуют о том, что параметры кристаллической ячейки Co3O4 для всех нанесенных катализаторов близки к литературным значениям 8.08 Å [41]. Размеры ОКР кобальтовой шпинели для катализаторов Cs/Ni-Co/носитель изменялись в узком интервале 220–270 Å для всех носителей. Для катализаторов Cs/Mg-Co/носитель размеры ОКР частиц кобальтовой шпинели в нанесенных катализаторах изменялись в более широком интервале 210–340 Å.
Величины удельной поверхности приготовленных образцов (SБЭТ) различаются и зависят от природы носителя (табл. 1). Низкими значениями SБЭТ (5–6 м2/г) обладают образцы на основе Mg2Al4Si5O18, средние значения (11–18 м2/г) демонстрируют образцы на основе α-Al2O3 и TiO2 и самые высокие значения – на основе CеO2 (45–50 м2/г).
Исследование катализаторов методом термопрограммируемой десорбции кислорода (O2-ТПД) показало, что десорбция также зависит от природы носителя (рис. 2, табл. 1). Согласно данным O2-ТПД, десорбция кислорода в образце 1%Cs/Co3O4 происходит при температуре 295°С. Авторы [42] считают, что слабосвязанная поверхностная форма кислорода Co3O4 удаляется в интервале 200–350°С, а выше 350°С происходит удаление кислорода из оксидной структуры. При модифицировании Co3O4 никелем или магнием формируются более слабосвязанные поверхностные формы кислорода, которые удаляются в интервале 70–200°С [37, 38, 43].
Профили десорбции кислорода для нанесенных на носители CeO2 и TiO2 катализаторов характеризуются наличием двух низкотемпературных пиков, что указывает на существование двух форм поверхностного кислорода: низкотемпературной (56 и 94°С) и высокотемпературной (258 и 300°С), соответственно (рис. 2). Согласно данным [33], чистый CeO2 также характеризуется наличием двух форм кислорода, удаление которых происходит при температурах выше 100 и 200°С. Десорбция кислорода для чистого TiO2 – рутил [44] должна происходить при температуре выше 350°С. Для приготовленных нанесенных катализаторов наблюдаются более низкие, чем для носителей TiO2 и CeO2, температуры десорбции в двух наблюдаемых низкотемпературных формах кислорода, и, напротив, более высокая температура десорбции кислорода относительно активного компонента во втором низкотемпературном пике десорбции, что, по-видимому, обусловлено взаимным модифицированием носителя и активного компонента. Действительно, известно, например, что модифицирование оксида церия катионами кобальта приводит к усилению окислительно-восстановительных свойств [42]. Таким образом, наблюдаемые для катализаторов на носителях TiO2 и CeO2 при температуре более 250°С пики десорбции обусловлены, наиболее вероятно, десорбцией с модифицированного носителем активного компонента и модифицированного активным компонентом носителя.
У катализаторов, нанесенных на высокотемпературные носители α-Al2O3 и Mg2Al4Si5O18, первый пик десорбции (в области температур 70–250°С) отсутствует, и соответственно отсутствует наиболее низкотемпературная поверхностная форма кислорода, относящаяся в случае носителей TiO2 и CeO2 к носителю. Действительно, согласно данным термопрограмированного изотопного обмена кислорода Martin и Dupre [45], начальные температуры обмена кислорода (в скобках) для α-Al2O3 (460°С) и SiO2 (650°С) достаточно высоки, что не позволяет протекать десорбции в области низких температур. Десорбция кислорода во втором пике протекает при температурах, меньших характерных для этих носителей, и более высоких, чем для оксида кобальта, по-видимому, вследствие модифицирования нанесенной кобальтовой шпинели. Таким образом, наблюдаемые для катализаторов на носителях α-Al2O3 и Mg2Al4Si5O18 при температуре более 300°С пики десорбции относятся, наиболее вероятно, к десорбции с модифицированного носителем активного компонента. Можно отметить, что количество десорбируемого кислорода в целом в ~2 раза выше для катализаторов, модифицированных магнием. Рассчитанные данные по количеству десорбированного кислорода представлены в табл. 1. Наибольшее количество десорбируемого кислорода ожидаемо демонстрируют катализаторы на носителе СеО2.
Выполненные исследования каталитической активности катализаторов показали, что величины для чистых активных компонентов выше, чем для нанесенных катализаторов (рис. 3, табл. 1). Это может быть обусловлено как меньшим количеством активного компонента в нанесенных катализаторах, составляющим ~50 мас. %, так и влиянием модифицирования активного компонента носителем. Среди нанесенных катализаторов активность зависит от носителя и изменяется для исследуемых составов активного компонента в ряду носителей следующим образом: (без носителя) > α-Al2O3 > CeO2 ≅ Mg2Al4Si5O18 > TiO2. Наблюдаемый ряд активности не коррелирует с величинами удельной поверхности образцов, что может свидетельствовать о различном характере модифицирования активного компонента в катализаторах. При этом скорость реакции, нормированная на единицу поверхности катализаторов, изменяется в ряду: α-Al2O3 > Mg2Al4Si5O18 ≅ (без носителя) > CeO2 ≅ TiO2, что указывает на меньшее модифицирование активного компонента в случае высокотемпературных носителей с низкой удельной поверхностью (корунд и кордиерит) и более значимое – в случае носителей с более высокой удельной поверхностью (СеO2 и TiO2). Авторы [46] указывают, что Co3O4 может частично внедряться в структуру рутила – TiO2 после прокаливания при 500°С. Оксид церия также способен взаимодействовать с Co3O4 и образовывать смешанные растворы различного состава, а в процессе термического взаимодействия структура CeO2 может претерпевать изменения, которые влияют на дисперсность частиц, что может приводить к экранированию нанесенного активного компонента [47] и сильно влиять на удельную активность (скорость реакции, нормированная на единицу поверхности).
Таким образом, высокотемпературные носители (α-Al2O3, Mg2Al4Si5O18) с низкой удельной поверхностью в меньшей степени модифицируют и блокируют активный компонент и поэтому более привлекательны, чем носители на основе CeO2 и TiO2, несмотря на присутствие в катализаторах, нанесенных на CeO2 и TiO2, наиболее слабосвязанной поверхностной формы кислорода, которая, по-видимому, обусловлена модифицированным носителем и в условиях реакционной среды, возможно, десорбируется.
Удельные каталитические активности (нормированная на единицу поверхности скорость каталитической реакции) активного компонента и нанесенных катализаторов в случае кордиерита оказались близки, а в случае корунда даже выше (рис. 3), несмотря на модифицирование активного компонента, возможно, вследствие более высокой степени использования активного компонента в нанесенных катализаторах (так что активность, нормированная на единицу массы активного компонента, в нанесенном катализаторе выше, чем в ненанесенном оксиде). В случае носителей CeO2 и TiO2 удельная каталитическая активность существенно ниже, что согласуется с предположением о более сильном модифицировании активного компонента в этих катализаторах и возможной блокировкой поверхности активного компонента частицами носителя.
Можно отметить отсутствие единой корреляции между активностью катализаторов и данными О2-ТПД по температурам и количеству десорбируемого кислорода, обычно обсуждаемой в литературе, в том числе и для модифицированных оксидов, для которых такая корреляция наблюдается [37, 38]. Отсутствие корреляции может означать, что скорость определяющей стадией каталитической реакции является стадия образования адсорбированного на поверхности кислорода, зависящая от подвижности поверхностного кислорода, а не стадия десорбции кислорода, определяемая прочностью связи [31]. Таким образом, при нанесении активного компонента на носители вследствие модифицирования активного компонента носителем происходит снижение подвижности кислорода на его поверхности, что может приводить к изменению скорость определяющей стадии процесса [26, 48, 49].
В условиях производств азотной и адипиновой кислот помимо закиси азота в отходящих газах присутствует кислород и пары воды [25], которые могут оказывать ингибирующее действие на катализатор. Поэтому все нанесенные образцы были исследованы в реакции разложения закиси азота на стабильность в присутствии этих ингибиторов при 300°С в течение 30 ч. В присутствии O2 (рис. 4а) нанесенные катализаторы демонстрируют стабильную работу лишь с незначительной потерей активности. В присутствии Н2О (рис. 4б) скорость дезактивации зависела не только от природы носителя, но и от природы активного компонента. Более активные катализаторы были менее стабильны, чем менее активные. Ряд потери активности для Ni-содержащих образцов: α-Al2O3 (63%) > TiO2 (52%) > Mg2Al4Si5O18 (47%) > CeO2 (22%) и для Mg-содержащих образцов: TiO2 (61%) > > CeO2(40%) ≥ Mg2Al4Si5O18 (39%) > α-Al2O3 (36%), в скобках указан процент потери активности по истечении 30 ч.
Активные компоненты без носителя, как и нанесенные катализаторы, значительно сильнее подвержены дезактивации в присутствии паров H2O, нежели кислорода. При этом нанесение активного компонента на любой из носителей увеличивает его устойчивость и снижает скорость дезактивации. Так, активный компонент 1%Cs/Mg0.1Co2.9O4 теряет 80% активности уже в первые 5 ч работы в присутствии паров H2O в реакционной смеси [37], в то время как нанесенные катализаторы работают гораздо стабильнее (рис. 4).
Таким образом, катализатор Cs/Mg–Co/α-Al2O3 является не только наиболее активным среди приготовленных нанесенных катализаторов, но и сохраняющим свою более высокую активность после 30-часовых испытаний в присутствии паров воды.
ЗАКЛЮЧЕНИЕ
Осаждением активных компонентов на носители CeO2, TiO2, Mg2Al4Si5O18, α-Al2O3 приготовлены катализаторы 1%Cs/Me0.1Co2.9O4 (Me = Ni, Mg)/носитель, содержащие ~50% активного компонента, представляющего собой твердый раствор со структурой шпинели на основе Со3О4.
Показано, что при нанесении наблюдаемое ранее различие в активности активных компонентов нивелируется, при этом максимальной активностью (температура достижения 50%-й степени превращения, скорость реакции, нормированная на величину удельной поверхности) характеризуются образцы, приготовленные на корундовом носителе, вероятно, вследствие меньшего модифицирования активного компонента носителем.
Обнаруженное отсутствие корреляции активности нанесенных катализаторов в реакции низкотемпературного разложения закиси азота и температурой начала десорбции кислорода или количеством десорбирующегося кислорода, вероятно, обусловлено изменением скорость-определяющей стадии процесса десорбции, вызванным модифицированием, что требует дополнительных исследований.
Показано, что ингибирующий эффект O2 и H2O на каталитическую активность зависит как от природы носителя, так и от состава активного компонента. Наиболее активным и сохраняющим более высокую активность в присутствии ингибиторов является катализатор 1%Cs/Mg0.1 Co2.9O4/корунд.
Данная работа выполнена в рамках государственного задания ФГБУН ИК СО РАН.
Список литературы
Pérez-Ramirez J., Kapteijn F., Schöffel K., Moulijn J.A. // Applied Catalysis. 2003. V. 44. P. 117.
Isupova L.A., Ivanova Y.A. // Kinetics and Catalysis. 2019. V. 60. № 6. P. 744.
Sadykov V.A., Isupova L.A., Zolotarskii I.A. et al. // Applied Catalysis A: General. 2000. V. 204. P. 59.
Исупова Л.А., Садыков В.А., Снегуренко О.И. и др. // Патент. 1998. № RU2117528C1.
Isupova L.A., Sadykov V.A., Snegurenko O.I. et al. / Пaтeнт. 1992. № US6489264B.
Isupova L.A., Sadykov V.A., Snegurenko O.I. et al. // Пaтeнт. 1997. № WO9840160A1.
Isupova L.A., Sadykov V.A., Tikhov S.F. et al. // Proc. of the 8 th Int. Symp. On Heterog. Catalysis, Varna, Bulgaria, 1996. V. 1. P. 479.
Isupova L.A., Sadykov V.A., Tikhov S.F. et al. // Catalysis Today. 1996. V. 27(1-2). P. 249.
Isupova L.A., Sadykov V.A., Tikhov S.F. et al. // Proc. 1st World Congress on Envir. Catal. Pisa. Italy: Environmental Catalysis for a better World and Life. 1995. P. 167.
Садыков В.А., Бруштейн Е.А., Исупова Л.А. и др. // Хим. пром-сть. 1997. № 12. С. 33.
Sadykov V.A., Isupova L.A., Tikhov S.F. et al. // Prep. of 16th World Petroleum Congress. Calgary. 2000. № 4. P. 9.
Носков А.С., Золотарский И.А., Кузьмин В.А. и др. Способ конверсии аммиака // Патент. 2000. № RU2145936C1.
Neumann J., Isupova L., Pinaeva L et al. // Patent. 2007. № WO2007/104403 (A1).
Jantzen S., Neumann J., Novel A. // Uhde Fertiliser Symposium, Dortmund, Germany. 2006. P. 17.
Pinaeva L.G., Prosvirin I.P., Dovlitova L.S. et al. // Catal. Sci. Technol. 2016. V. 6. P. 2150.
Пинаева Л.Г., Довлитова Л.С., Исупова Л.А. // Кинетика и катализ. 2017. Т. 58. № 2. С. 183.
Пинаева Л.Г., Исупова Л.А., Куликовская Н.А., Марчук А.А. Катализатор, способ его приготовления и способ разложения закиси азота // Патент. 2011. № РФ 2430781.
Пинаева Л.Г., Сутормина Е.Ф., Исупова Л.А. и др. Катализатор, способ его приготовления и способ окисления аммиака // Патент. 2011. № РФ2430782.
Чумаченко В.А., Исупова Л.А., Иванова Ю.А. и др. // Химия в интересах устойчивого развития. 2020. № 28. С. 210.
Grzybek T., Papp H. // Applied Catalysis. 1992. V. 1. P. 1271.
Groves M.C.E., Sasonow A. // Journal of Integrative Environmental Sciences. 2010. V. 7. № S1. P. 211.
Shen Q., Wang M., Wu H. et al. // Applied Surface Science. 2018. V. 441. P. 474.
Obalová L., Jiratova K., Karaskova K., Chromcakova Z. // Catalysis Today. 2012. V. 191. P. 116.
Wójcik S., Ercolino G., Gajewska M. et al. // Chemical Engineering Journal. 2019. V. 377. P. 120088.
Klegova A., Inayat A., Indyka P. et al. // Applied Catalysis B: Environmental. 2019. V. 255. P. 117745.
Исупова Л.А., Иванова Ю.А. // Кинетика и катализ. 2019. Т. 60. № 6. С. 725.
Konsolakis M. //ACS Catalysis_Revised MS. 2015. V. 5. № 11. P. 6397.
Ohnishi C., Asano K., Iwamoto S. et al. // Applied Catalysis B: Invironmental. 2007. V. 120. P. 145.
Dou Z., Zhang H., Pan Y., Xu X. / Journal of Fuel Chemistry and Technology. 2014. V. 42(2). P. 238.
Kapteijn F., Rodriguez-Mirasol J., Moulijn J.A. // Appl. Catal. B. Environ. 1996. V. 9. P. 25.
Ivanov D.V., Sadovskaya E.M., Pinaeva L.G., Isupova L.A. // J. Catalysis. 2009. V. 267. P. 5.
Pacultova K., Karaskova K., Strakosova J. et al. // C. R. Chimie. 2015. V. 18. P. 1114.
Grzybek G., Wójcik S., Ciura K. et al. // Applied Catalysis B: Environmental. 2017. V. 210. P. 34.
Pietrogiacomi D., Campa M.C., Carbone L.R. et al. // Applied Catalysis B: Environmental. 2016. V. 187. P. 218.
ójcik S., Ercolino G., Gajewska M. et al. // Chemical Engineering Journal. 2019. V. 377. P. 120088
Shen Q., Li L., Li J. et al. // Journal of Hazardous Materials. 2009. V. 163. P. 1332.
Иванова Ю.А., Сутормина Е.Ф., Исупова Л.А., Рогов В.А. // Кинетика и катализ. 2020. Т. 61. № 4. С. 577.
Иванова Ю.А., Сутормина Е.Ф., Исупова Л.А., Рогов В.А. // Кинетика и катализ. 2018. Т. 59. № 3. С. 365.
Исупова Л.А., Сутормина Е.Ф., Марчук А.А. и др. Носитель для катализаторов на основе оксида алюминия и способ его приготовления // Патент. 2016. № РФ 2623436.
Исупова Л.А., Куликовская Н.А., Марчук А.А. и др. Каталитический элемент для гетерогенных высокотемпературных реакций // Патент. 2018. № РФ 2693454.
Shaheen W.M., Ali A.A. // Materials Research Bulletin. 2001. V. 36. P. 1703.
Lu S., Wang F., Chen C. et al. // Journal of Rare Earths 2017. V. 35. № 9. P. 867.
Yan L., Ren T., Wang X. et al. // Catalysis Comm. 2003. V. 4. P. 505.
Liraa E., Huoa P., Hansena J.Ø. et al. // Catalysis Today. 2012. V. 182. P. 25.
Martin D., Duprez D. // J. Phys. Chem. B. 1997. V. 101. P. 4428.
Zhang A.-Y., He Y.-Y., Chen Y.-P. et al. // Chemical Engineering Journal. 2018. V. 334. P. 1430.
Balducci G., Fornasiero P., Di Monte R. et al. Kagpar J., Meriani S. // Catalysis Letters. 1995. V. 33. P. 193.
Ivanov D.V., Pinaeva L.G., Isupova L.A. et al. // Applied Catalysis A: General. 2013. V. 457. P. 42.
Ivanov D.V., Pinaeva L.G., Sadovskaya E.M., Isupova L.A. // Journal of Molecular Catalysis A: Chemical 2016. V. 412. P. 34.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии