

Журнал физической химии, 2021, T. 95, № 3, стр. 312-323
Физикохимия водных флюидов – основа технологических процессов с их участием
a Российская академия наук, Федеральный исследовательский центр химической физики им. Н.Н. Семенова
Москва, Россия
* E-mail: mysinev@yandex.ru
Поступила в редакцию 04.09.2020
После доработки 04.09.2020
Принята к публикации 24.09.2020
Аннотация
Рассмотрены некоторые физико-химические свойства водных флюидов (ВФ), лежащие в основе их практического использования, и критерии, позволяющие разграничить области параметров, в которых существуют качественные различия их физического состояния. Проанализирована качественная связь между типом и интенсивностью межмолекулярных взаимодействий в различных веществах и их макроскопическими характеристиками – температурами нормального кипения и критической точки. Особое внимание уделено плотности как макроскопическому параметру, отражающему влияние межмолекулярных взаимодействий в ВФ. Показано, что при всей технологической важности изобарных зависимостей для понимания механизмов воздействия различных факторов на свойства флюидов необходимо рассматривать изохорные и изотермические зависимости, особенно при анализе диэлектрических свойств воды и ее диссоциации на противоионы.
Исследование физикохимии процессов в газах при высоких давлениях и создание технологий на их основе имеют в нашей стране богатые традиции. Прежде всего, необходимо указать работы академика В.Н. Ипатьева [1], заложившего основы нефтехимии в России и существенно повлиявшего на ее развитие в США. В 1918 г. была создана Центральная химическая лаборатория ВСНХ СССР, давшая начало НИФХИ им. Л.Я. Карпова, а в 1931 г. – Государственный институт азота, впоследствии – ГИАП. Эти две мощнейшие исследовательские организации сыграли ключевую роль в создании и становлении не только азотной, но и в целом – химической промышленности. Нельзя не вспомнить таких выдающихся исследователей, как М.И. Темкин, И.Р. Кричевский, Д.С. Циклис и многих других, внесших огромный вклад в понимание состояния вещества, механизма и кинетики процессов, адсорбции и гетерогенных равновесий, в технику эксперимента при высоких давлениях [2–10]. Без этого создание производств таких важнейших и крупнотоннажных продуктов, как аммиак, метанол, карбамид было бы невозможным. В течение десятилетий указанные направления развивались также в ряде академических и прикладных институтов, в вузах разных регионов бывшего СССР. Многие из этих работ обобщены в фундаментальных монографиях и справочниках, давно не переиздававшихся и ставших библиографической редкостью [11–15].
Можно с уверенностью считать, что в течение продолжительного времени отечественные исследования в области физической химии, как теперь принято говорить, флюидного состояния находились на самых передовых позициях в мире. Созданы методы исследования, анализа и описания свойств веществ и их смесей в широком диапазоне параметров состояния [2, 4–8, 10–13]; открыт ряд ранее неизвестных явлений, наиболее яркое из них, по-видимому, существование области ограниченного смешения газов (расслоения газовых смесей) [4, 9].
Драматические изменения в стране в 1980–90-е годы привели к оскудению многих научных направлений и фактическому исчезновению технологической базы химической промышленности, что в полной мере коснулось и рассматриваемой области. Это совпало со временем формирования в мире отдельного направления, связанного с широким использованием свойств околокритических жидкостей и плотных газов для самых разных практических приложений, что вызвало бурное развитие исследований флюидного состояния веществ, в том числе фундаментального характера. Организовывались конференции, создавались специализированные издания (Journal of Molecular Liquids, Journal of Supercritical Fluids, Fluid Phase Equilibria, Supercritical Fluid Science and Technology, …), выделялись целевые гранты, формировалось сотрудничество между лабораториями.
В этих условиях возник целый ряд компаний, специализирующихся на производстве оборудования для исследований при высоких давлениях различного уровня (от лабораторного до пилотного) и различной степени оснащенности средствами контроля и анализа процессов – от довольно простых автоклавов для экстракции до реакторов различной конфигурации и систем микронизации, ректификации, флюидной хроматографии.
Отдельного упоминания заслуживает мероприятие, проходившее под эгидой НАТО в г. Кемер (Турция) с 18 по 31 июля 1993 г., – “Институт перспективных исследований сверхкритических флюидов – основы для приложений” (Advanced Study Institute on Supercritical Fluids – Fundamentals for Applications). В устоявшейся русскоязычной терминологии его следовало бы называть школой. По словам организаторов, их задача “… состояла в том, чтобы собрать вместе экспертов в данной области для критического обзора текущего состояния знаний и последних научных достижений в экспериментальных и теоретических методах”. По их убеждению (с которым невозможно не согласиться), приступающий к разработке технологий в этой сфере “… инженер должен быть знаком с широкой областью науки, простирающейся от фундаментальных физических теорий межмолекулярного взаимодействия и критических явлений до инженерных дисциплин, 〈теории〉 фазового поведения, моделирования термодинамических и транспортных свойств смесей флюидов, теории химической активности и кинетики, вплоть до 〈техники〉 экспериментов в незнакомой сильно сжимаемой среде” [16]. Прочитанные ведущими специалистами лекции легли в основу фундаментального издания, которое можно назвать энциклопедией по теории флюидного состояния и его практическим применениям [17]. Участниками школы стали 97 человек из 17 стран, включая некоторые части бывшего СССР.
Следует с сожалением констатировать, что по большому счету отечественные исследования и разработки оказались далеко в стороне от этих событий и бурного развития флюидных технологий. Хотя и нельзя считать, что в России в это время подобные исследования полностью прекратились. Оставались отдельные группы и лаборатории в организациях различного профиля и подчинения, которые либо продолжали уже начатые исследования, либо включались в них, несмотря на крайне неблагоприятные условия. Однако все это было весьма далеко от описанной выше систематической работы, которая велась в данном направлении за рубежом.
Очевидно, что продвижение в новых, технически сложных и, по сути, комплексных междисциплинарных направлениях возможно только в том случае, когда имеется конкретный заказ – со стороны промышленности или государственных институтов, заинтересованных в ускоренном развитии и получении конкретных значимых результатов. К сожалению, такая заинтересованность в нашей стране до сих пор не сформировалась. В таких условиях, к счастью, началось движение “снизу”, т.е. со стороны исследователей и технических специалистов, понимавших огромный потенциал флюидных технологий.
История самоорганизации “сверхкритического сообщества” еще ждет своего летописца (индивидуального или коллективного). Здесь же необходимо отметить особо человека, без которого невозможно было бы прийти даже к тем относительно скромным результатам, которые мы имеем на сегодня. Речь идет о Валерии Васильевиче Лунине, академике РАН, многолетнем руководителе Химического факультета МГУ, ставшем безусловным и непререкаемым лидером российского сообщества специалистов в области сверхкритических и смежных исследований и технологий. Вместе с другими соратниками Валерия Васильевича автору этих строк уже довелось писать о той роли, которую он сыграл в “собирании” разрозненных и зачастую разнонаправленных сил российских “сверхкритиков”, в организации школ и конференций, в становлении единственного отечественного специализированного журнала по сверхкритическим (СК) флюидам [18]. Последним по времени крупным организационным проектом, который Валерий Васильевич реализовал вместе с соратниками по МГУ (проф. В.Н. Баграташвили, д.х.н. Е.Н. Голубевой, к.х.н. О.О. Паренаго и другими) стало создание Центра по сверхкритическим флюидам. Под его эгидой велись и продолжаются исследования, организован общегородской семинар по СК-флюидам. Подробный рассказ обо всем этом требует иного формата. В настоящей работе хотелось бы осветить несколько иное – одно из важнейших научных направлений, которое было в центре внимания Валерия Васильевича. Речь идет о свойствах водных флюидов и процессах с их участием.
Интересно отметить два важных, и, казалось бы, не связанных между собой факта. Первый из них относится к уже упомянутой Школе в Кемере по СК-флюидам. Одной из главных тем, заявленных на ней, была химия с упором на процессы в водных средах [16]. Второй факт имеет прямое отношение к роли В.В. Лунина в активизации исследований по СК-флюидам в России. В 2005 г. был опубликован его (совместно с А.А. Галкиным) обзор “Вода в суб- и сверхкритическом состояниях – универсальная среда для осуществления химических реакций” [19], который до сих пор остается одной из наиболее цитируемых работ в российском “сегменте” литературы по СК- флюидам. Такой интерес к водным флюидам не случаен: вода обладает набором уникальных свойств, качественно отличающих ее от других веществ и при этом, как никакое другое вещество, способна менять эти свойства при изменении параметров состояния. Принципиально важно и то, что вода, пожалуй, самый “зеленый” и одновременно самый доступный растворитель, может служить средой и участником многочисленных процессов, а водные высокотемпературные флюиды играли и играют колоссальную роль в эволюции земной коры.
Приходится с горечью констатировать, что среди авторов фундаментального труда [16] нет ни одного нашего соотечественника, а из 208 цитированных в обзоре [19] источников лишь три русскоязычных и еще девять работ опубликовано отечественными авторами в англоязычных изданиях. Это, к сожалению, довольно точно отражает вклад российских исследователей в развитие данной области науки технологии в последние годы.
Интерес В.В. Лунина к этой тематике далеко не случаен. В течение многих лет в руководимой им Лаборатории катализа и газовой электрохимии (КГЭ) Химического факультета МГУ велись и продолжаются исследования в области синтеза и модифицирования оксидов в среде водных флюидов (см., например, [20–22]). Гидротермальный синтез оксидных систем (в том числе цеолитных) – катализаторов многочисленных процессов химии и нефтехимии – одно из главных направлений в Лаборатории кинетики и катализа (см., например, [23–25]), входящей в состав Кафедры физической химии, которой также руководил Валерий Васильевич. К этим работам он всегда относился с большим вниманием и интересом. Наконец, в период подготовки обзора [19] в Лаборатории КГЭ под его непосредственным руководством была создана установка и начинались работы по синтезу оксидных систем в СК-воде в проточных условиях.
Число работ, посвященных свойствам водных флюидов и самым разным процессам в них, огромно. Имеются многочисленные базы данных и обзоры, посвященные их свойствам и применениям (см., например, [26, 27]). Тем не менее, можно с уверенностью считать, что их потенциал в качестве среды, реагента и катализатора не только не исчерпан, но даже не до конца оценен. Причина этого очевидна – уникальность физико-химических свойств воды, ее структуры и их зависимости от параметров состояния. Не повторяя всего изложенного в [19], следует, тем не менее, подчеркнуть и уточнить некоторые из затронутых там аспектов темы и, возможно, дополнить их. Этому был посвящен доклад автора настоящей работы на заседании упомянутого семинара на базе Центра по сверхкритическим флюидам МГУ 20 февраля 2020 г.– последнем при жизни Валерия Васильевича и прошедшем уже, к сожалению, без его непосредственного участия. Главной своей задачей автор считал определение “зоны ответственности” науки о СК-флюидах применительно к соответствующим технологиям. В докладе освещались следующие основные вопросы:
1) особенности СК-флюидного и примыкающих к нему по параметрам состояний вещества;
2) сопоставление свойств водных флюидов с другими средами, в том числе используемыми в СК- и родственных технологиях, анализ физических причин различий между ними;
3) определение свойств флюидов, непосредственно влияющих на протекание в их среде и/или с их участием процессов различных типов;
4) обсуждение некоторых новых данных, позволяющих уточнить механизмы процессов, интересных с практической точки зрения.
Некоторые положения этого выступления изложены ниже.
Особенности сверхкритического флюидного состояния вещества
Сама по себе постановка вопроса, вынесенная в подзаголовок, могла бы показаться странной для обсуждения на специализированном семинаре. Однако характер дискуссий, которые зачастую возникают в печати и на конференциях по СК-флюидам, показывает настоятельную необходимость в явном виде рассмотреть некоторые важные положения для того, чтобы участники диалога могли надеяться на взаимопонимание, по крайней мере, по его предмету и содержанию. Довольно подробно данная тема обсуждена в недавней публикации автора [28], поэтому здесь будет дано лишь краткое резюме из проведенного анализа.
Если за основу при определении физического состояния флюида взять его агрегатное состояние, то важнейшим параметром следует считать сжимаемость – способность изменять плотность или удельный объем при изменении внешнего давления. Эта способность может характеризоваться критерием, включающим производную плотности по давлению (дифференциальная характеристика)
или параметром, называемым собственно сжимаемостью и непосредственно определяемым из термического уравнения состояния где ρ – плотность флюида, P – давление, R – универсальная газовая постоянная, T – температура, μ – молекулярная масса вещества, Vm – мольный объем.Можно легко показать, что для идеального газа (свободное движение невзаимодействующих частиц, занимающих суммарно малый объем по сравнению со свободным объемом системы) ζ = 1 и Z = 1, что следует из уравнения Клапейрона–Менделеева. Для идеальной несжимаемой жидкости (Vm, ρ не зависят от P) ζ = 0; при постоянной температуре Z ~ P, а при постоянном давлении Z ~ 1/T. Зависимость величины ζ от параметров состояния для воды подробно обсуждается в работе [28]. Изотермы Z(P) для воды в диапазоне 100–700°С, построенные по данным [29], приведены на рис. 1. Как следует из этих данных, при температурах до ~250–300°С параметр Z для паровой фазы (малые давления, верхняя/левая ветвь кривых) отличается от значения, характерного для идеального газа (Z = 1) не более, чем на 20–25%. При дальнейшем возрастании температуры отклонение от этого значения при приближении к кривой насыщения (ей соответствует разрыв кривых на рис. 1) становится все более значительным, что свидетельствует о нарастании неидеальности паровой фазы.
Рис. 1.
Зависимости параметра Z от давления при 100 (1), 150 (2), 250 (3), 300 (4), 350 (5), 374 (6), 400 (7), 450 (8), 500 (9), 600 (10) и 700°С (11) в натуральных (а) и двойных логарифмических (б) координатах.
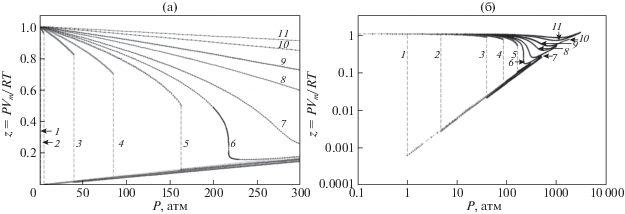
Следует отметить, что параметр Z менее чувствителен по сравнению с дифференциальным параметром ζ к нарастанию сжимаемости жидкой фазы при приближении в этом диапазоне температур к кривой насыщения справа, т.е. из области высоких давлений: трудно заметить отклонение от линейности зависимости Z от P почти вплоть до достижения критической точки.
После достижения критической температуры поведение параметров, описывающих сжимаемость водного флюида, резко отличается от того, что соответствует моделям идеального газа и несжимаемой жидкости. Диапазон параметров состояния, в котором такое поведение проявляется, можно было бы назвать областью существования сверхкритического водного флюида в собственном смысле слова.
При дальнейшем росте температуры неидеальность поведения, связанного со сжимаемостью вырождается, и уже выше температур 600–700°С при технически достижимых давлениях водные флюиды ведут себя во многом как идеальный газ.
Микроскопические причины такого поведения флюидов кратко рассмотрены в следующем разделе. Здесь же уместно сделать два уточняющих замечания.
Во-первых, строго говоря, резко неидеальное поведение СК-флюида при варьировании параметров состояния во многом следует тенденциям, которые нарастают в свойствах жидкой воды и плотного пара при приближении к кривой насыщения в области температур >300°С. Отсутствие возможности наблюдения таких изменений при более низкой температуре связано с конденсацией пара при достижении уже относительно небольших плотностей, т.е. не с отсутствием влияния межмолекулярных сил на свойства флюидов, а, наоборот, с их более сильным проявлением при относительно низких температурах. И принципиально важным отличием областей до критической точки и после нее является то, что до критической температуры рост плотности паровой фазы при повышении давления приводит к конденсации, уже невозможной при более высоких температурах. Это свидетельствует о снижении вклада межмолекулярных взаимодействий в свойства и поведение флюидов с ростом температуры.
Во-вторых, величина давления, при которой возникает резкая неидеальность, может быть значительно ниже давления в критической точке (217.75 атм по данным [29]). Это свидетельствует о том, что определение границ существования СК-флюида, особенно для смесей веществ, требует достаточно скрупулезного анализа, при котором плотность и ее зависимость от давления – существенно более важные параметры, чем само давление. С одной стороны, физически состояние СК-флюида может достигаться при давлениях ниже критического, но с другой – смесь веществ может не попадать в эту область и при давлениях существенно выше давления в критической точке даже для компонента, присутствующего в максимальной концентрации.
В [30] проанализировано поведение параметров Z и ζ при варьировании температуры и плотности воды, определены пределы и предложено уточнить терминологию для обозначения областей существования состояний водных флюидов (докртическая вода, субкритическая вода, разреженный пар, плотный пар, водный СК-флюид, область идеально-газового состояния), в свойствах которых имеются качественные различия, важные для реализации процессов различных типов.
Межмолекулярные взаимодействия и макроскопические свойства флюидов
Излишне указывать, что именно особенности физических и химических свойств водных флюидов во многом определяют возможности их практического использования. В свою очередь сами эти особенности определяются строением и свойствами молекулы воды и, в существенной мере, взаимодействиями между молекулами. В работах Л.П. Филиппова с соавт. [12, 31] проанализировано влияние формы потенциала межчастичного взаимодействия на макроскопические параметры веществ и показано, что именно подобие формы этого потенциала определяет подобие термодинамических свойств. На основе такого анализа были разработаны методы предсказания свойств веществ по их структурной формуле и алгоритмы подбора веществ с заданными свойствами для конкретных применений [13, 32]. Важно подчеркнуть, однако, что эти подходы хорошо работают в случае веществ, называемых Филипповым “нормальными” [12], к которым относятся неполярные соединения (например, инертные газы, молекулярный азот, углеводороды). Для таких веществ потенциал (энергия) межмолекулярного взаимодействия U(r) хорошо описывается простым уравнением Леннард-Джонса [33] (их даже называют “леннард-джонсовыми флюидами”):
илиСмысл параметров уравнения в обеих эквивалентных формах ясен из рис. 2: ε – максимальная энергия взаимодействия между молекулами в точке равновесия сил притяжения и отталкивания; r0 – равновесное расстояние между центрами молекул (2r0 ≅ d, где d – газокинетический диаметр молекулы); σ – расстояние между центрами молекул, на котором энергия взаимодействия равна нулю. На рис. 2 также приведена зависимость силы взаимодействия между молекулами F(r), которая равна нулю в точке r0, где силы притяжения и отталкивания равны по величине, и положительная ветвь которой (притяжение) имеет максимум в точке перегиба кривой U(r).
Рис. 2.
Потенциал Леннард-Джонса – зависимости энергии U(r) (1) и силы F(r) (2) взаимодействия между молекулами от расстояния между ними.
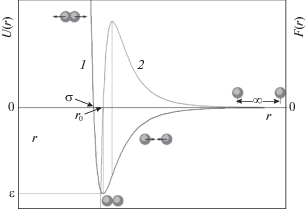
Показатель степени “6” у r во втором члене уравнения Леннард-Джонса, выражающем энергию притяжения, имеет строгое физическое обоснование: энергии всех трех типов межмолекулярных взаимодействий, определяемых свойствами системы электронов молекулы (ориентационное, индукционное и дисперсионное), обратно пропорциональны r6. Что касается показателя степени “12” при r в первом члене, то он не имеет теоретического обоснования, а лишь показывает, что энергия отталкивания возрастает гораздо быстрее, чем притяжение, при r → 0.
Строго говоря, уравнение Леннард-Джонса применимо к неполярным веществам, в молекулах которых электронная плотность имеет центрально-симметричное распределение. Однако оно очень наглядно показывает зависимость энергии взаимодействия между молекулами от расстояния и линейные масштабы, на которых могут проявляться основные качественные эффекты в поведении веществ.
Наличие дипольного момента у молекул приводит к необходимости введения дополнительных членов в описание потенциала взаимодействия. Важно также отметить, что при наличии дипольного момента у молекул в их взаимодействиях возникают выделенные направления, что не позволяет уже изобразить потенциал в виде графика в плоской системе координат.
Все еще более усложняется в случае так называемых ассоциированных веществ [12], для которых существенно влияние водородных связей. Эта группа включает, например, спирты и ряд неорганических соединений, в число которых входит, безусловно, и вода. К ним, по свидетельству Л.П. Филиппова, неприменимы относительно простые модели, позволяющие предсказывать термодинамические зависимости без привлечения данных о критических параметрах [12].
Вода, безусловно, относится именно к этой категории. Описание U(r) для нее содержит дополнительные члены, учитывающие, как минимум, электростатический вклад в энергию взаимодействия. В частности, при расчетах для воды в конденсированных состояниях (жидкость, твердое) методами молекулярной динамики часто используются потенциалы группы TIP4P (“4-Point Transferable Intermolecular Potentials” – четырехточечные переносимые межмолекулярные потенциалы) [34]. Четыре “точки”, которые являются в данном случае “опорными”, это атомы кислорода (один) и водорода (два), а также так называемый “М-центр”, лежащий на биссектрисе угла H–O–H в плоскости, образованной центрами двух атомов водорода и атома кислорода, на расстоянии ~0.15–0.16 Å от атома кислорода. Имеются и иные подходы к построению потенциалов для описания свойств воды в различных условиях [35], анализ которых не входит в задачу данной работы. Важно подчеркнуть, однако, что на сегодня существуют модели, в разной степени основанные на теоретических соображениях и эмпирических подходах, позволяющие описывать макроскопические свойства водных флюидов в широком диапазоне изменения параметров состояния.
После краткой характеристики межмолекулярных взаимодействий посмотрим, как они отражаются на макроскопических свойствах веществ. Приводимые ниже данные, как правило, взяты из справочников [36–39] или рассчитаны на основании информации, содержащейся в базе данных [29].
В табл. 1 приведены параметры ряда веществ – инертных газов и изоэлектронных им соединений элементов 1–3 периодов с водородом; в таблицу внесены также параметры CO2 – наиболее востребованного на сегодня в СК-флюидных технологиях вещества. Анализ этих данных приводит к нескольким важным заключениям. Во-первых, при возрастании числа электронов в атомах инертных газов резко возрастает их способность конденсироваться – возрастают температуры нормального кипения Tкип и критической точки Tкр (т.е. температура, выше которой конденсация невозможна). Несмотря на то, что параллельно с ростом числа электронов возрастает масса атомов, которая также может вносить вклад в конденсируемость (ср. критические температуры 3He и 4He, а также изотопных молекул водорода), его нельзя считать доминирующим. Это доказывает обратная тенденция – снижение Tкр при замещении протия на дейтерий в молекулах гидридов. Анализ большого массива данных показывает [40], что данная закономерность – общая для гидридов элементов, начиная от углерода и выше, а также то, что в указанных случаях эффект от снижения интенсивности межмолекулярных взаимодействий у дейтерозамещенных веществ, безусловно, превалирует над вкладом от увеличения массы молекул. Иными словами, даже в случае предельно неполярных веществ вклад электронных взаимодействий (дисперсионных) в макроскопические свойства будет основным.
Таблица 1.
Параметры молекул и характерные температуры некоторых веществ (N – число электронов в молекуле, µ – дипольный момент)
| Вещество | N | µ, D | Tкип, K (при 1 атм) | Tкр, K | Tкр/Tкип |
|---|---|---|---|---|---|
| 3He | 2 | 0 | 3.19 | 3.35 | 1.05 |
| 4He | 2 | 0 | 4.2 | 5.26 | 1.25 |
| H2 | 2 | 0 | 20.39 | 32.98 | 1.62 |
| HD | 2 | 0 | 22.13 | 35.91 | 1.62 |
| D2 | 2 | 0 | 22.92 | 38.35 | 1.67 |
| CH4 | 10 | 0 | 111.6 | 190.6 | 1.71 |
| NH3 | 10 | 1.46 | 239.8 | 405.7 (405.5) | 1.69 |
| H2O | 10 | 1.855 | 373.2 | 647.4 (644.7) | 1.73 |
| HF | 10 | 1.98 | 292.7 | 461.2 | 1.58 |
| Ne | 10 | 0 | 27.1 | 44.4 | 1.64 |
| SiH4 | 18 | 0 | 161.2 | 270 | 1.67 |
| PH3 | 18 | 0.58 | 185.2 | 324.2 (323.5) | 1.75 |
| H2S | 18 | 1.02 | 212.9 | 373.3 (372.2) | 1.75 |
| HCl | 18 | 1.08 | 188.1 | 324.2 (323.5) | 1.72 |
| Ar | 18 | 0 | 87.3 | 151.0 | 1.73 |
| CO2 | 22 | 0 | 194.7 | 304.2 | 1.56 |
Примечание. Образование водородных связей отмечено только для NH3, H2O и HF.
В скобках указаны критические температуры полностью дейтерированных соединений; величины Tкр веществ, для которых приведены параметры дейтерированных производных, даны по [40].
Второе важное заключение касается влияния полярности молекул на макроскопические свойства. На рис. 3 приведены зависимости критической температуры веществ от дипольного момента для 10- и 18-электронных молекул. Эти две зависимости качественно близки: для первых трех соединений (гидриды элементов групп IV–VI) наблюдается монотонный рост Tкр, который объясняется ростом полярности связей, дипольных моментов и соответственно энергии обусловленных ими взаимодействий. После этого происходит снижение ее величины для гидрида элемента VII группы. Указанная закономерность связана с числом полярных связей, присутствующих в молекуле: халькогениды водорода, имеющие в молекуле две полярные связи, имеют больше возможностей дипольного связывания между собой, чем молекулы галогенидов.
Рис. 3.
Зависимости от величины дипольного момента критической температуры молекул гидридов элементов II (кривая 1) и III периодов (кривая 2) и отношения Tкр/Tкип для них (прерывистая линия 3).
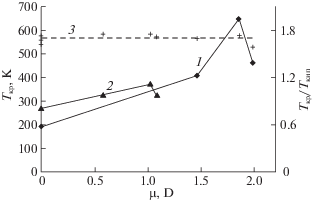
Третье наблюдение напрямую связано с водородными связями. Рисунок 3 наглядно показывает, что все значения Tкр для гидридов элементов III периода лежат между ее значениями для метана и аммиака – гидридов соседних элементов II периода. Причина в том, что молекулы метана – полностью симметричные, образованные элементами с близкими значениями электроотрицательности и содержащие 10 электронов – имеют на восемь электронов меньше, чем молекулы моносилана при близких геометрии и соотношении электроотрицательностей (хотя и с обратным знаком) образующих его атомов. Соответственно энергия взаимодействия между молекулами последнего, обусловленная дисперсионными силами, существенно выше. Однако уже молекулы аммиака связаны в конденсированном состоянии более прочно, чем молекулы не только фосфина, но и сероводорода. Это обусловлено не только более высокой электроотрицательностью атома азота по сравнению с серой (и соответственно более выраженными зарядами на атомах в молекуле и интенсивностью ориентационных взаимодействий), но и способностью к образованию водородных связей, которая практически не выражена у гидридов элементов III периода. При дальнейшем росте полярности связей “элемент–водород” энергия водородных связей монотонно возрастает в ряду NH3 < H2O < HF. Однако это не приводит к росту Tкр у HF по сравнению с водой. Главным фактором здесь служит наличие в молекуле H2O двух протонированных атомов водорода и двух неподеленных пар электронов у атомов кислорода. В результате, несмотря на то, что энергия каждой водородной связи во фтористом водороде выше, чем в воде, его молекулы могут образовывать лишь цепочки типа H–F···H–F···H–F, в то время как молекулы воды способны формировать тетраэдрические структуры – трехмерные сетки, в которых каждая молекула образует с другими вплоть до четырех водородных связей. Этим и обусловлены как рекордное для гидридов значение Tкр, (647.1 K по данным [29]), так и многие другие аномальные свойства воды.
Наконец, еще одна закономерность, которая прослеживается в данных табл. 1, связана с отношением Tкр/Tкип. Несмотря на значительные различия в характеристиках молекул, о чем шла речь выше, это отношение практически постоянно для всех приведенных веществ за исключением гелия и лежит в пределах 1.56–1.75. Причем, как видно из табл. 1 и рис. 3, никакой закономерности в вариациях указанного отношения в зависимости от строения и иных особенностей веществ не прослеживается. Если следовать подходам, развитым в [12, 13], это дает основания полагать, что будут работоспособными и иные корреляции, общие даже для столь разнородных веществ, как инертные газы и ассоциированные флюиды, включая воду, поскольку обе точки (нормального кипения и критическая) принадлежат кривой насыщения – линии на “P–T”-диаграмме состояния, на которой сосуществуют равновесные жидкая и паровая фазы.
Намеченная в последней части монографии [12] “исследовательская программа” продолжает оставаться актуальной, как в части создания теории термодинамического подобия для многокомпонентных систем (смесей, растворов), так и в отношении распространения развитых ее автором подходов, позволяющих связать особенности потенциалов взаимодействия с макроскопическим поведением, на ассоциированные вещества, включая воду. То, что данная программа имеет хорошие перспективы реализации, показывает относительно простой пример.
Выше отмечалось, в сколь сильной степени макроскопическое поведение определяется характером и интенсивностью межмолекулярных взаимодействий. В этом смысле инертные газы (максимально неполярные вещества, типичные леннард-джонсоновские флюиды) и вода (вещество, способное существовать в форме максимально ассоциированного флюида) представляют собой две противоположные крайности. Из табл. 1 видно, в какой степени различны их характеристики. Так, температуры нормального кипения и критической точки различаются на ~500 К, а их отношение – более чем в 4 раза.
На рис. 4а представлены зависимости критерия ζ от давления для аргона и воды при равных значениях приведенной температуры τ = T/Tкр = 1.05. Физические температуры при этом весьма различны – 158.2 К для аргона и 679.5 К для воды. Видно, что в области низких давлений состояние обоих веществ близко к идеально-газовому (ζ → 1 при P → 0), а с ростом давления сжимаемость резко возрастает, что характерно для состояния СК-флюида. Ход этих зависимостей, однако, совершенно различен. Если же воспользоваться хорошо известной процедурой замены физических величин на их приведенные значения (π = P/Pкр, ρ* = ρ/ρкр) и построить зависимость величины ζ* (вычисленной с использованием приведенной плотности ρ* вместо физической плотности ρ), от ρ*, то, как видно на рис. 4б, можно получить почти совпадающие зависимости. Это показывает, что даже для столь различных по строению и свойствам молекул веществ подходы теории термодинамического подобия, в данном случае – метод соответственных состояний, вполне работоспособны. Как известно, указанный метод основан на тождественности уравнений состояния (например, уравнения Ван-дер-Ваальса), применимых для описания различных веществ. Анализ уравнений состояния, применимых к различным группам веществ и физических соображений, лежащих в их основе, может привести к дальнейшему продвижению в реализации “исследовательской программы”, сформулированной Л.П. Филипповым [12].
Рис. 4.
Зависимости величины дифференциального критерия ζ от давления (а) и приведенного критерия ζ* (см. текст) от приведенной плотности (б) для аргона (1) и воды (2).
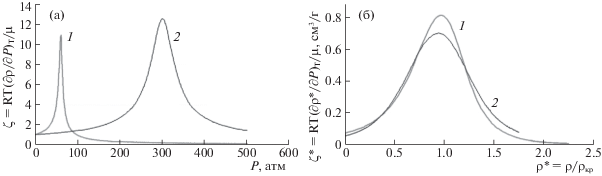
Резюмируя изложенное в данном разделе, важно еще раз подчеркнуть: существование области неидеальности макроскопических свойств в районе критической точки, как и возможность конденсации паровой фазы при более низких температурах, – следствие действия сил межмолекулярного взаимодействия. Резкое возрастание сжимаемости газо- или парообразных флюидов при возрастании их плотности и уменьшении мольного объема связано напрямую с тем, что расстояния между молекулами снижаются до величин, при которых ускоренно растет сила притяжения между ними (в области r < (2–3)r0 – см. рис. 2). Иными словами, при этих плотностях межмолекулярные силы притяжения действуют совместно и однонаправлено с внешним давлением. Сильная зависимость интенсивности взаимодействия между молекулами от плотности флюида приводит к высокой параметрической чувствительности многих их свойств, представляющих технологический интерес, в первую очередь – растворяющей способности. В сочетании с возможностью технически относительно простого варьирования плотности это делает флюидные технологии столь востребованными.
Свойства водных флюидов, определяющие технологический интерес к ним
Несмотря на очевидное преимущество воды как, пожалуй, самого “зеленого” растворителя, высокие параметры (температура, давление) ее критической точки делают ее значительно менее привлекательной по сравнению с другими флюидами для относительно простых технических приложений. Тем не менее, вода обладает рядом уникальных свойств, благодаря которым она остается весьма интересным и востребованным растворителем, реагентом, средой для проведения процессов различных типов. Главное ее отличие и преимущество – ионный характер водных сред и сильная зависимость диэлектрических свойств и ионной силы от параметров состояния. Именно этими свойствами водных флюидов определяется их способность селективно растворять и разделять вещества различной полярности, участвовать в реакциях, включающих стадии гидролиза и обратимой гидратации, играть роль катализатора кислотно-основных процессов.
Важна также способность воды вступать в реакции окислительно-восстановительного типа при достаточно высоких температурах. Однако эта способность, по-видимому, не имеет прямого отношения к физическому состоянию водных флюидов.
Если выше были рассмотрены свойства воды, которыми она принципиально не отличается от иных веществ, что доказывается применимостью к описанию состояния водных флюидов общих подходов, развитых в теории термодинамического подобия [12], то в данном разделе мы остановимся на уникальных свойствах воды как высокополярного вещества и ионной системы. Этим свойствам уделено пристальное внимание и в обзоре [19].
Однако здесь необходимо сделать важное уточнение: существует принципиальное различие в изменениях рассматриваемых свойств водных флюидов при движении вдоль изобары (т.е. при P = const) и вдоль изохоры (ρ = const). Технически и технологически (особенно для процессов, ведущихся в потоке) более приемлемо рассмотрение изобарных зависимостей, которые проще исследовать экспериментально. Техника измерения давления и поддержания его постоянства прекрасно отработана. Измерение и поддержание постоянства плотности – гораздо более сложная задача. Единственный надежный метод на сегодня – проведение процессов в автоклавах, однако и здесь существует ряд сложностей, неучет которых может приводить к существенным неопределенностям и даже ошибкам при проведении экспериментов и анализе их результатов [41]. Ситуация может измениться при более широком использовании методов непосредственного контроля плотности среды [42]; они, однако, пока не стали элементом стандартных экспериментальных методик. Так или иначе, сегодня рассмотрение изохорных свойств и поведения флюидных систем еще не стало общепринятым, хотя для специалистов, занимающихся анализом данных по свойствам флюидных систем, преимущества именно такого подхода совершенно очевидны (см., например, [43–45]).
При рассмотрении диэлектрических свойств и ионного поведения воды необходимость изохорного рассмотрения еще более очевидна, чем в случае анализа термодинамических и динамических свойств. И дело далеко не только в том, что при изохорном описании получаются более простые уравнения и зависимости [43, 44]. Совершенно очевидно, что многие свойства флюидных систем определяются соотношением энергий теплового движения (температура) и межмолекулярного взаимодействия. Интенсивность последнего, как уже отмечено, напрямую связана с плотностью, которая служит мерой количества вещества в единице объема и, следовательно, отражает расстояния между молекулами во флюиде. Если же мы рассматриваем диэлектрические свойства вещества, то они также зависят от числа элементарных диполей и поляризуемых частиц в единице объема, их взаимной ориентации (корреляции), т.е. опять-таки, от плотности. Для воды, как и прочих ассоциированных флюидов, существенное влияние на диэлектрические свойства оказывают водородные связи [44], среднее число которых на одну молекулу и время жизни существенно зависят от параметров состояния [46].
Пожалуй, еще более важным фактором плотность является при рассмотрении диссоциации воды на противоионы и на форму их существования в системе. Действительно, если мы рассмотрим процессы диссоциации воды и сольватации продуктов диссоциации (можно ограничиться протоном), то по закону действующих масс равновесие реакций:
(1)
${{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {{{\text{H}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}},$(2)
${{{\text{H}}}^{ + }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {{{\text{H}}}_{{\text{3}}}}{{{\text{O}}}^{ + }},$(3)
${{{\text{H}}}_{{\text{3}}}}{{{\text{O}}}^{ + }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {{{\text{H}}}_{{\text{5}}}}{\text{O}}_{2}^{ + }$и, следовательно, степень диссоциации воды, концентрации ионов, ионная сила системы зависят от концентрации воды, а она напрямую связана с плотностью водного флюида, так как для чистой воды ее молярная концентрация C практически эквивалентна плотности (с точностью до единиц измерения объема):
Если это не принимать во внимание, то возникает кажущийся парадокс: диссоциация воды – заведомо эндотермический процесс, однако степень диссоциации и концентрации противоионов с ростом температуры резко снижаются. Приводя такую зависимость авторы [19], конечно же, указывают, что она определена при постоянном давлении. Однако было бы весьма важно также подчеркнуть, что сложная форма зависимости ионного произведения (ИП) воды от температуры на изобаре есть следствие наложения трех зависимостей:
(а) собственно ИП от плотности/концентрации (закон действующих масс);
(б) констант равновесия реакций (1)–(3), в первую очередь, диссоциации, от температуры (уравнение Вант-Гоффа);
(в) плотности от давления (уравнение состояния флюида).
Из изохорного описания последний фактор – наиболее сложный и наименее определенный – исключается. Именно это и дало основание авторам [43] упростить зависимости при переходе от изобарных к изохорным. Здесь же хотелось более подчеркнуть физическую суть отличий двух описаний и причину, по которой они оказываются мнимо противоречивыми: убывающий характер изобарной зависимости ИП от температуры при том, что изохорная зависимость – возрастающая, определяется исключительно снижением плотности воды с ростом температуры при P = const.
В работе [43] проведен анализ экспериментальных данных по зависимостям ИП воды от различных параметров и получено уравнение, описывающее зависимость ионного произведения воды (пропорционального константе равновесия диссоциации) KW от температуры и плотности следующего вида:
(5)
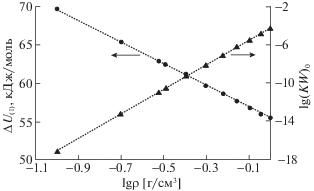
$\begin{gathered} \lg {{K}_{{\text{W}}}} = A + B{\text{/}}T + C{{T}^{2}} + D{\text{/}}{{T}^{3}} + \\ + \;(E + F{\text{/}}T + G{\text{/}}{{T}^{2}})\lg r. \\ \end{gathered} $Построенные по уравнению (5) и приведенным в [43] значениям его параметров зависимости приведены на рис. 5. Они показывают хорошее согласие температурной зависимости KW с уравнением Вант-Гоффа
или температурный коэффициент в котором связан с изменением внутренней энергии в реакции (1) ΔU(1), а величина предэкспонента lg(KW)0 – с изменением энтропии. Как показывает рис. 6, обе эти величины линейно изменяются с ростом плотности водного флюида, причем эндотермичность процесса снижается, а величина предэкспоненты растет. В результате обе зависимости KW – от температуры при постоянной плотности и от плотности при постоянной температуре – согласно уравнению (5) возрастают почти экспоненциально. И лишь наложение убывающей зависимости плотности от температуры при постоянном давлении приводит к падению ИП воды на изобаре [19].Рис. 5.
Температурные зависимости величины ионного произведения воды по уравнению (5) при различных плотностях водного флюида: 1 – 0.1, 2 – 0.2, 3 – 0.3, 4 – 0.4, 5 – 0.6, 6 – 0.8, 7 – 1.0 г/см3.
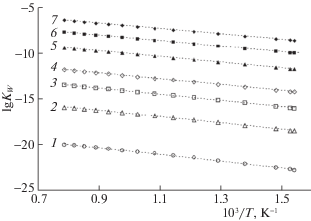
Важно подчеркнуть: на полученных в экспериментах зависимостях ИП воды от температуры и плотности, которые использованы при получении уравнения (5), не наблюдается никаких изломов, перегибов или иных особенностей, которые указывали бы на влияние каких-либо структурных перестроек в водных флюидах на диссоциацию молекул. По данным авторов [43], полученное описание работоспособно в широкой области параметров состояния: при температурах от тройной точки до 1000°С и давлениях от 1 до 104 атм.
В работе [44] проанализирован большой объем экспериментальных данных, а также различные теоретические подходы к описанию зависимости диэлектрической постоянной и связанных с ней свойств водных флюидов от параметров состояния. Как и в случае с диссоциацией воды, авторы указывают, что существенно более информативны зависимости от плотности, нежели от давления. В целом же, авторы вынуждены констатировать, что “Отсутствие прочной физической основы для формулирования поведения диэлектрической проницаемости воды и пара до боли очевидно, несмотря на длительные и сосредоточенные усилия некоторых из величайших умов в области физической химии”. Они также сетуют на большие противоречия в экспериментальных данных, включая сверхкритическую область.
Тем не менее, в результате скрупулезного анализа вопроса авторам удалось сформировать достаточно удобную для использования базу данных, включающую оптимизированное аналитическое описание и вычисленные на его основе табулированные зависимости статической диэлектрической постоянной воды в зависимости от температуры, давления и плотности, по которым построены кривые, приведенные на рис. 7. Они показывают, что эта величина монотонно возрастает при увеличении плотности флюида и снижается с ростом температуры. Как и в случае ионного произведения воды, на кривых нет изломов, перегибов или еще каких-либо особенностей.
Рис. 7.
Зависимости статической диэлектрической постоянной воды от плотности при постоянной температуре (а) и от температуры при постоянной плотности (б).
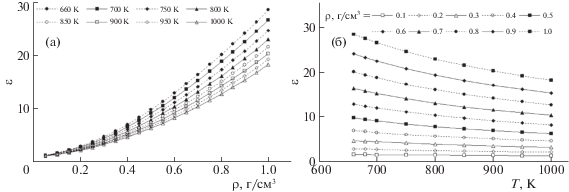
Следует подчеркнуть еще раз, что представленные на рис. 5–7 зависимости построены по аналитическим выражениям, которые, в свою очередь, получены обработкой больших массивов экспериментальных данных. Хотя это не есть результат прямых измерений свойств водных флюидов, их можно считать максимально надежными на данном этапе развития исследований.
Анализ данных, представленных в этом разделе, заставляет более внимательно оценивать получаемые зависимости скорости химических процессов, протекающих в водных флюидах, от их плотности. Ранее [47] при рассмотрении структурирования аморфного кремнезема в среде водных флюидов различной плотности мы обсуждали ее влияние исключительно в терминах кинетического закона действующих масс, т.е. с точки зрения зависимости скорости процесса от концентрации воды как реагента. Возможно, однако, в данном случае имеет определенное значение и изменение диэлектрических свойств и степени диссоциации воды (ионной силы среды). Разделение этих эффектов потребует отдельных исследований.
ЗАКЛЮЧЕНИЕ
Анализ изменения макроскопических свойств водных флюидов при изменении параметров состояния показывает, что
– ослабление влияния сил межмолекулярного взаимодействия на свойства жидкости возникает до достижения критической температуры в области, которую принято называть областью существования субкритичесокй воды;
– влияние сил межмолекулярного взаимодействия на свойства флюида быстро снижается с ростом температуры выше критической точки;
– давление –“плохой” параметр для характеристики состояния флюида; тенденции изменения свойств вдоль изобары могут вводить в заблуждение относительно их природы, особенно в районе критической точки и при T > Tкр;
– основные параметры, влияющие на состояние и свойства флюида, – плотность (количество вещества в единице объема, расстояние между молекулами, возможность определенных ориентаций и движений определенного характера, …) и температура; именно они определяют соотношение между силами взаимодействия молекул и интенсивностью теплового движения, которое выражается в макроскопических свойствах веществ;
– сильная зависимость разнородных свойств водных флюидов (термодинамических, динамических, диэлектрических, химических) от параметров состояния требует развития новых подходов к исследованиям влияния различных факторов на протекание процессов с их участием.
Список литературы
Ипатьев В.Н. Каталитические реакции при высоких температурах и давлениях: 1900–1933. М.-Л.: Изд-во АН СССР, 1936. 774 с.
Кричевский И.Р., Казарновский Я.С. // Журн. физ. химии. 1939. Т. 13. С. 378.
Кричевский И.Р., Гамбург Д.Ю. // Там же. 1943. Т. 17. № 4. С. 215.
Кричевский И.Р. Фазовые равновесия в растворах при высоких давлениях. М.-Л.: Госхимиздат, 1946. 120 с.
Кричевский И.Р. Термодинамика критических бесконечно разбавленных растворов. М.: Химия, 1975. 120 с.
Темкин М., Пыжев В. // Журн. физ. химии. 1939. Т. 13. № 7. С. 851.
Темкин М. // Там же. 1943. Т. 17. № 4. С. 269.
Темкин М. // Там же. 1943. Т. 17. № 5–6. С. 414.
Циклис Д.С. Расслоение газовых смесей. М.: Химия, 1969. 160 с.
Циклис Д.С. Техника физико-химических исследований при высоких и сверхвысоких давлениях. Изд. 4-е, пер. и доп. М.: Химия, 1976. 432 с.
Гоникберг М.Г. Химическое равновесие и скорость реакций при высоких давлениях. Изд. 2-е, М.: Изд-во АН СССР, 1960. 272 с.
Филиппов Л.П. Подобие свойств веществ. М.: Изд-во Моск. ун-та, 1978. 256 с.
Филиппов Л.П. Прогнозирование теплофизических свойств жидкостей и газов. М.: Энергоатомиздат, 1988. 166 с.
Варгафтик Н.Б. Справочник по теплофизическим свойствам газов и жидкостей. М.: Наука, 1972. 721 с.
Справочник азотчика (в 2-х томах) / Под ред. Е.Я. Мельникова. 2-е изд., М.: Химия, 1986-87. 512+464 с.
Kiran E., Levelt Sengers J.M.H. Preface; in Supercritical Fluids. Fundamentals for Application. / Ed. by E. Kiran, J.M.H. Levelt Sengers. NATO Science Series E: Applied Science, V. 273. Springer-Science+Business Media, B.V., 1994. p. IX–XII.
Supercritical Fluids. Fundamentals for Application / Ed. by E. Kiran, J.M.H. Levelt Sengers. NATO Science Series E: Applied Science, V. 273. Springer-Science+Business Media, B.V., 1994. 800 p.
“In Memoriam – Воспоминания о Валерии Васильевиче Лунине” // СКФ-ТП. 2020. Т. 15. № 1. С. 3.
Галкин А.А., Лунин В.В. // Успехи химии. 2005. Т. 74. № 1. 2005. С. 24.
Danchevskaya M.N., Ivakin Y.D., Torbin S.N. et al. // J. Mater. Sci. 2006. V. 41. № 5. P. 1385.
Danchevskaya M.N., Ivakin Y.D., Torbin S.N., Muravieva G.P. // J. Supercritical Fluids. 2008. V. 46, Iss. 3. P. 358.
Синев М.Ю., Ивакин Ю.Д., Шашкин Д.П. и др. // СКФ-ТП. 2019. Т. 14. № 3. С. 45.
Маерле А.А., Касьянов И.А., Московская И.Ф., Романовский Б.В. // Журн. физ. химии. 2016. Т. 90. № 6. С. 902.
Ivanova I.I., Kolyagin Y.G., Kasyanov I.A. et al. // Angewandte Chemie – International Edition, 2017. V. 56. № 48. P. 15344.
Шкуропатов А.В., Князева Е.Е., Пономарева О.А., Иванова И.И. // Нефтехимия. 2018. Т. 58. № 5. С. 529.
Brunner G. Hydrothermal and Supercritical Water Processes. Supercritical Fluid Science and Technology. Elsevier Book Series, V. 5, 1st ed., 2014. 604 p.
Near-Critical and Supercritical Water and Their Applications for Biorefineries / Ed. by Zh. Fang, Ch. Xu. Springer Science+Business Media, Dordrecht, 2014, 474 p.
Синев М.Ю., Шаповалова О.В. // СКФ-ТП. 2020. Т. 15. № 3. С. … . В печати.
NIST Chemistry WebBook, NIST Standard Reference Database Number 69, 2018 (https://webbook.nist.gov/chemistry/); https://doi.org/10.18434/T4D303.
Лагунова Е.А., Ивакин Ю.Д., Синев М.Ю. и др. // СКФ-ТП. 2019. Т. 14. № 4. С. 49.
Толстунов Д.А., Филиппов Л.П. // Журн. физ. химии. 1982. Т. 56. № 1. С. 129.
Охоцимский А.Д., Филиппов Л.П. // Докл. АН СССР. 1985. Т. 280. № 3. С. 60.
Jones J.E. // Proc. Roy. Soc. London. 1924. V. A 106. P. 463.
Abascal J.L.F., Vega C. // J. Chem. Phys. 2005. V. 123. P. 234505.
Cisneros G.A., Wikfeldt K.T., Ojamäe L. et al. // Chem. Rev. 2016. V. 116. № 13. P. 7501.
Справочник химика, т. 1. Л.–М.: Госхимиздат, 1963, 1072 с.
Карапетьянц М.X., Карапетьянц М.Л. Основные термодинамические константы неорганических и органических веществ. М.: Химия, 1968. 472 с.
Краткий справочник физико-химических величин / Под ред. К.П. Мищенко и А.А. Равделя. Изд. 7-е. Л.: Химия, 1974. 200 с.
Свойства неорганических соединений. Справочник / Под ред. В.А. Рабиновича. Л.: Химия, 1983. 392 с.
Рабинович И.Б. Влияние изотопии на физико-химические свойства жидкостей. М.: Наука, 1968. 308 с.
Синев М.Ю., Гордиенко Ю.А., Пономарева Е.А., Ивакин Ю.Д. // СКФ-ТП. 2019. Т. 14. № 2. С. 116.
Авдеев М.В., Баграташвили В.Н., Коновалов А.Н. и др. // Там же. 2007. Т. 2. № 1. С. 28.
Marshall W.L., Franck E.U. // J. Phys. Chem. Ref. Data. 1981. V. 10. № 2. P. 295.
Fernández D.P., Goodwin A.R.H., Lemmon E.W. et al. // Ibid. 1997. V. 26. № 4. P. 1125.
Абдулагатов И.М., Скрипов П.В. // СКФ-ТП. 2020. Т. 15. № 1. С. 34.
Горбатый Ю.Е. Влияние температуры и давления на ближний порядок в жидкой и надкритической воде: Дис. … докт. физ.-мат. наук, М.: ИХФ АН СССР, 1988 г. 301 с.
Лагунова Е.А., Ивакин Ю.Д., Синев М.Ю. и др. // СКФ-ТП. 2019. Т. 14. № 4. С. 49.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии