

Журнал физической химии, 2021, T. 95, № 3, стр. 324-341
Возможности молекулярного моделирования кинетических процессов в сверхкритических условиях
a Российская академия наук, Институт общей и неорганической химии им. Н.С. Курнакова
119991 Москва, Россия
* E-mail: tovbinyk@mail.ru
Поступила в редакцию 28.08.2020
После доработки 28.08.2020
Принята к публикации 31.08.2020
Аннотация
Рассмотрены современные возможности моделирования кинетики сверхкритических процессов на основе представлений теории абсолютных скоростей реакций для неидеальных реакционных систем, учитывающей межмолекулярные взаимодействия, которые меняют эффективную энергию активации элементарных стадий. Установлено, что это позволяет теории описывать скорости элементарных стадий для произвольных температур и плотностей реагентов в разных фазах. Рассмотрено применение данной теории в широкой области изменения параметров состояния (давления и температуры) при расчете элементарных бимолекулярных реакций и диссипативных коэффициентов в сверхкритических условиях. Приведены расчеты обобщенных зависимостей в рамках закона соответственных состояний для коэффициентов сжимаемости, вязкости и теплопроводности чистых веществ, а также для бинарных смесей (коэффициенты сжимаемости, самодиффузии, взаимной диффузии, и вязкости). Продемонстрировано влияние плотности и температуры на скорости элементарных стадий в сверхкритических условиях: эффективные энергии активации реакций, коэффициентов диффузии и сдвига, а также константы равновесия адсорбции. Обсуждены различия в моделях, использующих эффективные параметры моделей, и перспективы развития этих моделей за счет учета различий в размерах, вкладов колебательных движений компонентов, а также повышение точности описания эффектов корреляции.
Переход к сверхкритическим (СК) состояниям газовой смеси связан с повышением в системе температуры и давления [1–4]. К процессам, протекающим в СК-средах, относятся гомогенные химические реакции в объемных фазах и гетерогенные каталитические реакции, процессы создания новых материалов, в том числе, и наноразмерных частиц, экстракции, хроматографии, физико-химические процессы в пористых средах и т.д. СК-среды могут резко изменить условия протекания химических реакций по сравнению с нормальными условиями при давлении в одну атмосферу и температурах ниже критической. Это происходит за счет высокой плотности среды по сравнению с газовой фазой и быстрого теплоотвода. Изменения условий в реакционной системе смещает все химические равновесия и скорости реакций, что позволяет осуществлять такие процессы, которые при нормальных условиях маловероятны. На этом основан поиск новых физико-химических процессов, реализация которых обеспечит разработку более экологически чистых производств. Важность указанных процессов определяет необходимость разработки физико-химических и математических моделей процессов, связанных с использованием СК-флюидов (СКФ) в качестве среды, реагентов или растворителей. Варьирование давления в газе – один из путей управления общим ходом процесса. Одновременно в СК-процессах повышается температура, и оба фактора по-разному влияют на характеристики объемных и поверхностных процессов, в том числе процессов адсорбции на открытых поверхностях и внутри пористых материалов. Известно, что повышение температуры уменьшает величину адсорбции, тогда как увеличение давления увеличивает величину адсорбции.
В данной работе рассматриваются существующие возможности моделирования физико-химических процессов, которые реализуются в однородных СК-фазах на молекулярном уровне. Одна из главных проблем расчета скоростей реакций в конденсированных фазах заключается в учете влияния среды [5–11]. В конденсированной фазе молекулы реагентов постоянно находятся в поле действия соседних молекул. Потенциалы взаимодействия реагентов и соседних молекул влияют на потенциальную поверхность реакции, которая меняет свои характеристики по отношению к той же реакции в отсутствие соседей (как это имеет место в газовой фазе).
Скорость элементарной стадии бимолекулярной реакции рассчитывается на основании закона действующих масс [12–14]:
(1)
${{w}_{{ij}}} = {{k}_{{ij}}}{{n}_{i}}{{n}_{j}},\quad {{k}_{{ij}}} = {\text{ }}k_{{ij}}^{0}\exp ( - {{E}_{{ij}}}{\text{/}}{{k}_{{\text{B}}}}T),$Уравнения закона действующих масс предполагают, что реализуется равновесное распределение молекул в реакционной системе и лимитирует стадия химического превращения, т.е. предполагается отсутствие диффузионного торможения на макроскопическом уровне, диффузионно-контролируемого режима реакции на молекулярном уровне и, наконец, влияния межмолекулярных взаимодействий.
В плотных фазах межмолекулярные взаимодействия приводят к отклонениям от законов идеальных реакционных систем, поэтому рассматриваются неидеальные реакционные системы. Впервые влияние повышения давления относительно атмосферного на каталитический процесс синтеза аммиака было исследовано в работе М.И. Темкина [16] (см. также [17, 18]). В ней было показано, что неидеальность реакционной системы при повышенных давлениях приводит к изменению констант скоростей элементарных стадий.
В неидеальных реакционных системах основными выражениями для скорости элементарных стадий являются построенные в рамках ТАСР [12]. Для бимолекулярной стадии, вместо уравнения (1), скорость запишется как
где $k_{{ij}}^{*}$ – константы скоростей элементарных реакций, которые представляются в виде(3)
$k_{{ij}}^{*} = {{k}_{{ij}}}^{0}\frac{{{{\alpha }_{i}}{{\alpha }_{j}}}}{{\alpha _{{ij}}^{*}}}\exp ( - {{E}_{{ij}}}{\text{/}}{{k}_{{\text{B}}}}T){\text{ }} = {{k}_{{ij}}}{{\alpha }_{i}}{{\alpha }_{j}}{\text{/}}\alpha _{{ij}}^{*},$Из выражения (1) следует, что с повышением температуры скорость реакции экспоненциально увеличивается. Выражение (1) отражает влияние СКФ через произведение концентраций реагентов, если молекулы СКФ вступают в химическую реакцию с другими компонентами реакционной смеси, и никак не отражает влияние СКФ, если молекулы СКФ не вступают в реакцию. Однако при повышении давления даже в случае присутствия инертного СКФ скорость реакции уменьшается. Это уменьшение обусловлено тем, что: 1) СКФ занимает объем системы и “мешает” встрече реагентов и 2) латеральное притяжение между реагентами и СКФ создает вокруг каждого реагента ассоциат из молекул инертного вещества.
Реальный химический процесс всегда состоит из нескольких стадий, зависящих от механизма брутто-реакции и транспорта молекул. Знание концентрационных зависимостей транспортных коэффициентов также важно для моделирования общего СК-процесса в аппаратах непрерывного или периодического действия. Поэтому ниже обсуждается влияние межмолекулярных взаимодействий на коэффициенты самодиффузии и вязкости в однородных системах (объемной фазе или на однородной поверхности) при увеличении концентрации СКФ.
В общем случае при обсуждении влияния СКФ на брутто-процессы следует учитывать разные аспекты данной проблемы: 1) повышение давления (плотности) повышает роль латеральных взаимодействий; 2) повышение температуры смещает равновесные концентрации компонентов в реакционных системах, что может приводить к протеканию химических реакций, которые были маловероятны при низких температурах (например, термическая диссоциация воды, приводящая к значительному повышению величины ионного произведения) [1, 22]; 3) переход к СК-условиям меняет состояние материалов реакционной системы (например, под влиянием СКФ меняются состояние кокса на катализаторах или свойства полимерных матриц в мембранах). Все эти вопросы должны рассматриваться совместно, но главным среди них остается первый вопрос: как меняются скорости отдельных стадий, из которых формируются механизмы брутто-процессов, указанных в пунктах 2 и 3 [23]. От решения этого вопроса зависит точность описания брутто-процесса.
Описание свойств конденсированных фаз основано на учете межмолекулярных взаимодействий, поэтому необходимо иметь адекватную процедуру описания потенциалов межмолекулярного взаимодействия. В качестве сверхкритических растворителей используют молекулы воды, СО2, фреонов, низкомолекулярных алканов, спиртов и т.д.; процессы с участием таких растворителей протекают в многокомпонентных растворах. Поэтому в первую очередь необходимо уметь описывать свойства СКФ в объемной фазе и при контакте с поверхностями твердых пористых и непористых тел. Для непористых тел возможно влияние СКФ на поверхность, приводящее к его растворимости, например, в полимерных матрицах. Чтобы описать свойства этих флюидов и их разнообразных растворов необходимо учитывать области применения молекулярно-кинетической теории в рамках модели решеточного газа (МРГ) [10, 11], разработанной ранее для взаимодействующих компонентов смесей с короткодействующим сферическим потенциалом взаимодействия, а также переход на более сложные межмолекулярные потенциалы. Равновесные распределения компонентов в МРГ рассматривались ранее в работах [19–21, 24]. Эти потенциалы отражают форму многоатомных молекул, а также наличие направленных связей и многочастичных взаимодействий (специфические взаимодействия и поляризацию молекул).
ТЕОРИЯ НЕИДЕАЛЬНЫХ ХИМИЧЕСКИХ РЕАКЦИЙ
Для теоретического анализа используется ТАСР неидеальных реакционных систем в рамках МРГ, которая оперирует параметрами межмолекулярного взаимодействия [10, 11, 25, 26]. Будем учитывать латеральные взаимодействия между всеми ближайшими соседними молекулами (реагентами и СКФ) в квазихимическом приближении (КХП), которое описывает эффекты корреляции ближнего порядка. Напомним, что эта теория позволяет рассматривать весь диапазон плотностей флюидов от разреженного газа до жидкости и в широком диапазоне температур от тройной точки системы до температур, многократно превышающих критическую температуру. Данный подход обеспечивает получение самосогласованного описания равновесного состояния реакционной смеси и скоростей элементарных стадий, что дает возможность рассмотрения сложных многостадийных физико-химических процессов на основе моделей атомно-молекулярного уровня. Основные положения этой теории многократно публиковались, поэтому здесь для наглядности ограничимся уравнениями для скорости бимолекулярной реакции и уравнениями для равновесного распределения компонентов.
В МРГ объем системы разбивается на ячейки размером порядка среднего размера молекулы.
В МРГ любая ячейка может быть занята частицей сорта i , где 1 ≤ i ≤ s – 1 (s – число компонентов системы), или быть свободной (вакантной), в этом случае индекс
i = s. Как указано выше, ${{{\theta }}_{i}}$ – степень заполнения решеточной системы частицами сорта i (или ее числовая плотность), причем $\sum\nolimits_{j = 1}^s {{{{\theta }}_{i}}} = 1$, а θ – полное заполнение решеточной системы всеми компонентами системы i, 1 ≤ i ≤ ≤ s – 1, θ = $\sum\nolimits_{j = 1}^{s - 1} {{{{\theta }}_{i}}} $, т.е. доля свободных узлов равна ${{{\theta }}_{s}} = 1 - {\theta }$. Отношения 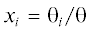 представляют собой мольную долю компонента i среди всех молекул смеси.
представляют собой мольную долю компонента i среди всех молекул смеси.
Каждая ячейка имеет z ближайших соседей, между которыми реализуется латеральное взаимодействие. Параметр этого взаимодействия между парами соседних частиц ij обозначим через εij. Параметр взаимодействия любой частицы с вакансией равен нулю. Межчастичные взаимодействия приводят к коррелированному пространственному распределению реагентов (вместо хаотического). Для описания этого факта необходимо использовать парные функции распределений частиц θij, характеризующие вероятность встречи реагентов, необходимой для реализации химического превращения, которая протекает на малых расстояниях между реагентами. Именно этот факт позволяет в задачах химической кинетики ограничиться, в первую очередь, рассмотрением пространственного распределения только ближайших соседей.
Выражение для скорости бимолекулярной реакции типа Ленгмюра–Хиншельвуда $w_{{fg}}^{{{\text{AB}}}}$, протекающей на двух соседних узлах f и g однородной системы между компонентами А + В, согласно теории неидеальных реакционных систем [25, 26], запишется следующим образом:
(4)
$w_{{fg}}^{{{\text{AB}}}} = k_{{fg}}^{{{\text{AB}}}}\exp ( - {\beta \varepsilon }_{{{\text{AB}}}}^{{}}){\theta }_{{fg}}^{{{\text{AB}}}}\Lambda _{{fg}}^{{{\text{AB}}}},$(5)
$\begin{gathered} \Lambda _{{fg}}^{{{\text{AB}}}} = \prod\limits_{h \in (z(f) - 1)} {S_{{fh}}^{{\text{A}}}} \prod\limits_{h \in (z(g) - 1)} {S_{{gh}}^{{\text{B}}}} , \\ S_{{fh}}^{{\text{A}}} = \sum\limits_{j = 1}^s {t_{{fh}}^{{{\text{A}}j}}} \exp ({\beta }({\varepsilon }_{{{\text{A}}j}}^{*} - {\varepsilon }_{{{\text{A}}j}}^{{}})). \\ \end{gathered} $В выражениях (4) и (5) функция $t_{{fh}}^{{ij}}$ = $\theta _{{fh}}^{{ij}}$/$\theta _{j}^{i}$ – условная вероятность нахождения частиц j рядом с частицами i, здесь функции $\theta _{{fh}}^{{ij}}$ имеют смысл вероятности нахождения рядом двух частиц i и j. Так как система однородна, то здесь нижние индексы, указывающие на номера соседних узлов, введены только для того, чтобы указать на различия в положениях реагентов: $\theta _{{fh}}^{{ij}}$ = θij и $t_{{fh}}^{{ij}}$ = tij. В обсуждаемом приближении kAB – константа скорости полагается постоянной и равной константе скорости в уравнении (1). В общем случае θij ≠ θiθj. В отсутствие взаимодействия молекул уравнения переходят в хорошо известные уравнения идеальных реакционных систем, для которых θij = θiθj.
Латеральные взаимодействия двояко влияют на скорости элементарных реакций. Они меняют: 1) вероятность встречи реагентов (в формуле (4) находится сомножитель θАВ вместо произведения θАθВ); и 2) высоты активационных барьеров по сравнению с идеальной системой (с газовой фазой) – этот вклад описывает функция неидеальности $\Lambda _{{fg}}^{{{\text{AB}}}}$. В функциях SА и SВ суммирование ведется по всем s сортам соседних частиц, обозначаемых индексом j. Функции Si представляют собой компоненты функции неидеальности реакционной системы, которые учитывают влияние соседних частиц на высоту активационного барьера реакции. Разность ${\delta }{{{\varepsilon }}_{{ij}}} = {\varepsilon }_{{ij}}^{*} - {{{\varepsilon }}_{{ij}}}$, присутствующая в формуле (5), определяет изменение энергии активации реакции A + B за счет влияния соседней частицы j, находящейся рядом с реагентом i, по сравнению с величиной энергии активации ЕАВ в формуле (1) для малых степеней заполнений (или для идеальной системы).
В химической кинетике для газовых фаз наибольшее внимание уделяется температурной зависимости скорости реакции [13, 14], которая изменяется по экспоненциальному закону, а концентрационная зависимость для бимолекулярной реакции – по квадратичному закону. Для неидеальных систем, как видно из выражений (4) и (5), в которых функция неидеальности состоит из произведения функций ${\text{exp}}({\beta \delta }{{{\varepsilon }}_{{ij}}})$ и ${{t}_{{ij}}}$, температурный и концентрационный факторы взаимосвязаны друг с другом, поэтому концентрационные зависимости становятся такими же важными, как и температурные.
Равновесное распределение молекул в однородной системе отвечает функциям θij, определяемым в квазихимическом приближении (КХП) из решения алгебраической системы уравнений совместно с условием нормировки:
(6)
${{{\theta }}_{{ij}}}{{{\theta }}_{{ss}}} = {{{\theta }}_{{is}}}{{{\theta }}_{{sj}}}\exp ( - {\beta }{{{\varepsilon }}_{{ij}}}),\quad \sum\limits_{j = 1}^s {{{{\theta }}_{{ij}}}} = {{{\theta }}_{i}}.$Для расчета бимолекулярной реакции на однородной поверхности следует учитывать, что поверхностные концентрации молекул, входящие в уравнения для скорости реакции, определяются из уравнений адсорбции основных реагентов А и В в присутствии СК-компонента С. Уравнения, описывающие адсорбцию смеси на однородной поверхности, записываются в КХП с учетом межчастичного взаимодействия как
(7)
${{a}_{i}}{{p}_{i}} = \left( {\frac{{{{{\theta }}_{i}}}}{{{{{\theta }}_{s}}}}} \right)S_{i}^{z},\quad {{S}_{i}} = 1 + \sum\limits_{j = 1}^{s - 1} {{{x}_{{ij}}}} {{t}_{{ij}}},$Наличие парных функции распределений частиц θij отражает эффекты корреляции и самосогласованность описания равновесия скоростей элементарных стадий и равновесия в системе. Если эффекты корреляции отсутствуют, то в системе нарушается данное самосогласование, и константы скоростей элементарных стадий в прямом и обратном направлениях не обеспечивают расчет константы локального равновесия [10, 11, 25, 26]. Корректность расчета элементарных стадий, в свою очередь, позволяет рассчитать все диссипативные коэффициенты при переносе импульса (коэффициент сдвиговой вязкости), энергии (коэффициент теплопроводности) и массы (коэффициенты самодиффузии и взаимной диффузии).
МОДЕЛЬ ЭФФЕКТИВНОГО ПАРНОГО ПОТЕНЦИАЛА
Учет только ближайших соседей на жесткой решетке сужает возможности количественного описания многих экспериментальных кривых даже в равновесии [19–21, 24]. В работе [27] было расширено использование МРГ за счет учета внутренних движений частицы внутри ячеек, что привело к интегральным уравнениям [28] как в теории жидкого состояния [29–32], однако, сохранилось условие невозможности множественного заполнения ячеек разными частицами как в МРГ. Это позволило получить континуальное КХП, которое упрощается за счет учета свойств системы [33], если использовать характерные решеточные построения, включающие концепции об исключенном объеме для центра масс частиц в ячейке, о мягкости решеточной структуры и колебаниях соседних молекул.
Эти построения непосредственно связаны с разными степенями свободы молекул [27]. Так, поступательные движения молекул реализуются в “свободном” объеме; “мягкость” решеточной структуры определяется средней величиной относительного смещения молекул, а колебательные движения молекул реализуются в их связанном состоянии. Эти понятия о разных типах движений четко определены в разреженном газе и твердой фазе. В плотных газах и жидкостях все молекулы постоянно находятся в поле друг друга, и понятия о “чистых” движениях имеют условный смысл.
“Исключенный” объем [33]. В плотных фазах соседние молекулы препятствуют движению произвольно выбранной центральной частицы, в результате чего часть объема системы оказывается ей недоступна. Влияние соседей на величину исключенного объема легко пояснить на примере свободной и заполненной решетки. В первом случае центр массы молекулы может находиться в любой точке ячейки; во втором – только в малой центральной части ячейки, так как остальной объем блокирован соседями, фиксированными в центрах своих ячеек. Величину этого объема V(θ ~ ~ 1) = κ3, доступного для движения центра массы центральной молекулы можно определить из простой геометрической модели, где κ – среднеквадратичное смещение частиц в твердом теле в гармоническом приближении для температур выше температуры Дебая. Cреднеквадратичное смещение выражается через параметры леннард-джонсовского взаимодействия без привлечения дополнительных подгоночных параметров [33]. Отношение V(θ)/V(θ = 0) = L(θ)d, где L(θ) – линейный размер области, доступный центру масс молекулы в данной ячейке, характеризует долю свободного объема, в которой движется центр массы молекулы. В самом простейшем приближении L(θ) = tAv + κtAA, где tAA – условная вероятность нахождения рядом двух молекул. Более точно исключенный объем аппроксимируется в рамках геометрической модели, фиксирующей положения соседних молекул [33].
Мягкость решетки характеризует среднее расстояние λ между узлами (средний размер ячейки), которое определяется из минимума свободной энергии системы. Для газообразного состояния размер ячейки связан с расстоянием молекул в изолированном димере. Для твердого тела средний размер ячейки определяется взаимодействиями молекулы со всеми окружающими соседями. Для определения среднего размера ячейки λ не используется никаких дополнительных параметров. Он находится из условия минимума свободной энергии системы [34] или через теорему вириала [35, 36]. В результате с ростом плотности происходит уменьшение размера ячейки. Эти изменения играют важную роль при количественном расчете фазовых диаграмм, так как меняется положение жидкофазной ветви (относительно такой же ветви для жесткой решетки), которая дает плотность тройной точки ρ ~ 0.81–0.86 (в зависимости от z), что близко к экспериментальному значению плотности в тройной точке и к результатам численного моделирования методом молекулярной динамики.
Колебания молекул характеризуют их тепловое движение в связанном состоянии. Колебания влияют на средние значения межмолекулярных взаимодействий и, соответственно, через условие мягкости решетки на все равновесные распределения. Учет малых отклонений молекул при их колебаниях относительно среднего размера ячейки (с учетом ангармонических вкладов эта техника была разработана в [37–39]) приводит к появлению температурной зависимости параметра латеральных взаимодействий: ε = ε0(1 – uT), где функция u аппроксимирует вклад колебательного движения молекул. Факт температурной зависимости энергетического параметра хорошо известен для решеточных моделей [19–21, 40–42], когда функция u рассматривалась в качестве подгоночного параметра. Подход [27] позволяет связать эту функцию с потенциальными атом-атомными кривыми без введения каких-либо параметров.
Учет указанных молекулярных свойств решеточной системы резко упрощает аппроксимацию континуального распределения молекул, сохраняя традиционную технику решения системы алгебраических уравнений в МРГ, что уменьшает время расчета равновесного распределения молекул до двух-трех порядков по сравнению с использованием интегральных уравнений.
Приложения к паро-жидкостным системам. При описании экспериментальных данных в широком диапазоне плотностей важную роль играет тип межмолекулярного потенциала. Для расчета газообразных и жидких фаз наиболее часто используется леннард-джонсовский потенциал с параметрами: σ (диаметр твердой сферы) и ε0 (глубина потенциальной ямы изолированного димера) [43]. Эти параметры были получены из экспериментальных данных о втором вириальном коэффициенте и коэффициенте сдвиговой вязкости при низких плотностях, и их традиционно используют в методах Монте-Карло и молекулярной динамики при любых плотностях. Однако, хорошо известно, что для высоких плотностей необходим учет вклада тройных взаимодействий [31, 43–45]. Для работы во всем диапазоне плотностей в МРГ была построена следующая аппроксимация эффективного парного потенциала типа Леннард-Джонса [46–48]:
(8)
$\begin{gathered} {{\varepsilon }_{{{\text{AA}}}}}(r) = 4{{\varepsilon }_{{{\text{ef}}}}}\left( {{{{\left( {\frac{{\sigma {\text{ }}}}{{\text{r}}}} \right)}}^{{{\text{12}}}}} - {{{\left( {\frac{{\sigma {\text{ }}}}{{\text{r}}}} \right)}}^{{\text{6}}}}} \right), \\ {{\varepsilon }_{{{\text{ef}}}}} = {{\varepsilon }_{0}}(1--{{d}_{{{\text{tr}}}}}{{\Delta }_{{{\text{1r}}}}}{{t}_{{{\text{AA}}}}})\left( {1--uT} \right), \\ \end{gathered} $Возможности указанных выше модификаций МРГ для описания экспериментальных данных иллюстрируются на примере плотных газов аргона (σ = 0.34 нм и ε0 = 119 K [43]) в объемной фазе на рис. 1а. На поле сопоставлены расчеты фактора сжимаемости Z = р/nкТ, где n – числовая плотность, в МРГ [46] и по вириальному разложению [51] с экспериментальными данными при температурах T = 162 K [52] (кривая 2). Видно, что для описания экспериментальных данных и для достижения согласия с решеточной моделью необходимо использовать как минимум пять членов вириального разложения. На вставке показана кривая расслаивания аргона, рассчитанная с учетом калибровки (см. ниже) и температурной зависимости (8) и экспериментальные данные [53]. Плотность аргона отложена в нормированном виде ρ/ρc, где ρc – плотность в критической точке. Во всей области температур имеется удовлетворительное согласие. Критический показатель β для рассчитанной и экспериментальной кривых равен 0.37.
Рис. 1.
а – Концентрационные зависимости фактора сжимаемости аргона [46]; эксперимент при 162 К [52] (кружки), расчет по МРГ (кривая 1); расчет по вириальным уравнениям [51]: 2 – учет второго вириального коэффициента, 3 – учет второго и третьего вириальных коэффициентов, 4 – учет со второго по четвертый вириальных коэффициентов; на вставке: фазовая диаграмма аргона, значки – экспериментальные значения [53], сплошная линия – расчет [50] при dt = 0.15, u = 0.00075; б – фазовые диаграммы Ar, N2 и смеси Ar–N2 при различных составах с мольной долей азота xN2 = 0 (1), 0.2 (2), 0.4 (3), 0.6 (4), 0.8 (5), 1.0 (6); эксперимент [54] – значки, расчет – сплошные линии [55].
На рис. 1б показано применение обсуждаемой модели к расчету кривых расслаивания бинарной смеси Ar–N2 при различных составах с различной мольной долей азота [54].
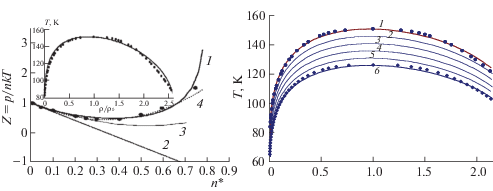
Теория дает удовлетворительное согласие с экспериментом и для других газов [56]. Для плотных газов, подчиняющихся “закону” соответственных состояний (молекулы которых не обладают специфическими взаимодействиями) имеет место так называемый обобщенный коэффициент сжимаемости [43, 51, 57]: в координатах Z – “приведенное” давление для разных “приведенных” температур все вещества имеют одни и те же кривые (“приведенные” величины нормированы на соответствующие значения в критических точках). На рис. 2 приведены обобщенные коэффициенты сжимаемости в широком диапазоне температур. Видно, что основные особенности коэффициента сжимаемости простейших газов для разных температур удовлетворительно отражаются в МРГ до 1000 атм [47]. Рассчитанные кривые [47] с точностью до 3–4% описывают обобщенный коэффициент сжимаемости, построенный по экспериментальным данным [43, 51, 57]. Важная особенность обобщенных зависимостей: если компоненты смеси не нарушают применимость закона соответственных состояний (как это показано на рис. 1б), то эти зависимости могут применяться для оценок и для смесей [57].
Рис. 2.
Обобщенные величины фактора сжимаемости плотных газов, подчиняющихся закону соответственных состояний [56].
Рассмотренная обобщенная величина фактора сжимаемости плотных газов, подчиняющихся закону соответственных состояний включает в себя критическую область [56]. На основе идей скейлинговой теории [58–60] в работах [28, 61] был предложен приближенный способ расчета равновесных распределений вблизи критической области с помощью калибровочных функций. Суть этого способа заключается в возможности разделения вкладов близкодействующего потенциала и влияния крупномасштабных флуктуаций, которые не зависят от локальных свойств потенциальных функций. Калибровочная функция отражает вклады крупномасштабных флуктуаций по отношению к вкладам от близкодействующих эффектов. Более подробно о калибровочной функции см. также [62–64].
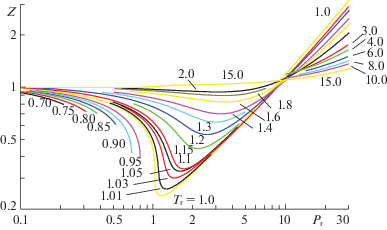
Также приведем пример использования полученных параметров межчастичного взаимодействия для расчета равновесных характеристик смесей при высоких давлениях. Возможность используемой модели для описания расслоения газов в объемной фазе при высоких давлениях продемонстрирована на рис. 3. Расчет достаточно удовлетворительно соотносится с экспериментальными данными для широкого диапазона изменения параметров состояния. В расчете использовались традиционные представления о законе соответственных состояний компонентов смеси, хотя аммиак несколько выпадает из области применимости данного закона. С увеличением давления этот закон нарушается, тем не менее, данный пример показывает, что используемый подход может быть применен и в случае отклонений от закона соответственных состояний с целью получения качественных результатов.
Рис. 3.
Зависимости давления системы от концентрации аммиака в бинарной смеси аммиак–азот при различных температурах (эксперимент [65] – слева, расчет – справа).
Влияние СКФ на характеристики адсорбционных процессов. Повышение давления в газовой фазе при протекании поверхностных процессов (адсорбционных, каталитических, мембранных и т.д.) влияет как на скорости элементарных стадий с участием газовых компонентов реакционной системы, так и на их условия равновесия [1–3]. В общем случае такое влияние можно разделить на два случая в зависимости от состава газовой смеси. Будем относить к первому случаю газовую смесь, содержащую молекулы, которые хорошо адсорбируются и принимают участие в поверхностных процессах. Повышение давления в системе непосредственно увеличивает концентрацию адсорбированных частиц (реагентов). Классический пример такой системы – процесс синтеза аммиака [16], протекающего при давлениях в несколько сотен атмосфер.
Ко второму случаю следует относить газовую смесь, содержащую в большом количестве молекулы, которые слабо адсорбируются или практически не адсорбируются (инертные компоненты). Повышение давления также непосредственно увеличивает степень заполнения основных реагентов, которые хорошо адсорбируются, но одновременно “принуждает” плохо адсорбирующиеся молекул также увеличивать свою степень заполнения. Если количество плохо адсорбирующихся молекул в газовой фазе значительно превышает количество основных реагентов, то их степени заполнений могут быть соизмеримы, и на этом может быть основана возможность более мягкого управления протеканием процесса. К этому типу систем относятся многие полимерные системы, взаимодействие которых, особенно, со сверхкритическим диоксидом углерода (СК‑СО2) активно исследуются [66, 67]. Повышенное внимание объясняется тем, что СК‑СО2 служит растворителем или пластификатором для целого ряда полимеров, обладая при этом такими преимуществами в сравнении с традиционными растворителями, как низкая стоимость, экологическая чистота, негорючесть, легкость удаления из полимера после проведения процесса и другие (см. ниже).
В работе [68] показано, что главный фактор влияния на характеристики адсорбционных процессов на однородной поверхности – концентрация СКФ, одного из компонентов адсорбирующейся смеси, и его влияние определяется конкурирующим соотношением энергий связи между адсорбентом и основным компонентом и СКФ. Анализ проведен как для физической, так и для химической адсорбции в зависимости от степени заполнения поверхности. Методы численного исследования адсорбционных систем изложены в [10, 64].
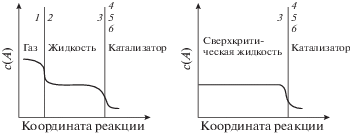
Приповерхностная область. Для поверхностных процессов важную роль играют распределения адсорбирующихся компонентов и реагентов вблизи поверхности в СК-условиях. Эти распределения схематически обычно представляются (рис. 4) в виде профилей фаз по нормали к поверхности адсорбента или катализатора [1].
Молекулярная теория дает возможность рассчитать детальные молекулярные распределения компонентов реакционных смесей (реагентов и СКФ-растворителя) в поле твердого тела. На рис. 5 представлены типичные равновесные распределения (профили плотности) смеси адсорбата и СКФ вблизи адсорбента в СК-условиях. Расчеты сделаны при фиксированном значении количества основного компонента θА = 0.1 (кривые 1) и варьировании доли СКФ (компонент С – кривые 2). Правая колонка относится к θС = 0.1, а левая – к θС = 0.5. В каждом ряду указаны температура Т и межчастичные параметры взаимодействия (в кал/моль). Варьируется энергия взаимодействия компонентов смеси со стенкой: первый ряд относится к слабому взаимодействию, второй к промежуточному и третий ряд – к сильному взаимодействию. Значения номера q меняются от первого монослоя до 11. Видно, что основное изменение плотности происходит на расстояниях шестого монослоя. Предельные значения плотности вдали от поверхности отвечают общей мольной доле компонента.
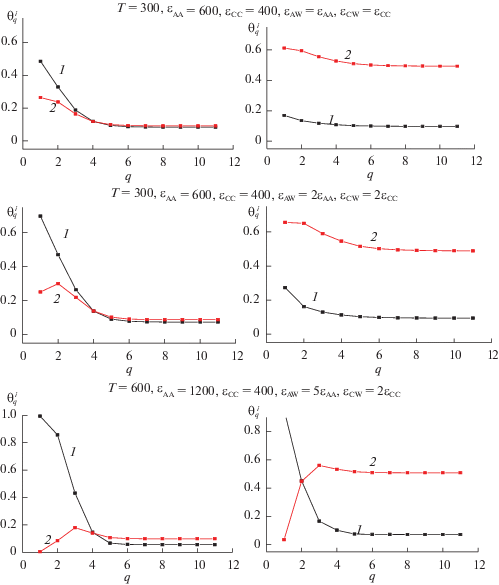
Кривые на рис. 5 показывают, что схематическое представление [1] в целом соответствует молекулярным расчетам, хотя детали таких распределений могут существенно отличаться. Однако при низких температурах схематические представления о жидкой прослойке для СКФ-систем в общем случае могут не отвечать молекулярным распределениям. Анализ показал, что двукратное повышение температуры при слабых энергиях взаимодействия молекул со стенкой оказывает заметное влияние на концентрационный профиль, тогда как при сильных межчастичных взаимодействиях вариация температуры не может существенно изменить этот профиль. Такое поведение коррелирует с анализом влияния СКФ на сорбционные и кинетические характеристики в объемной фазе. В целом, молекулярная теория позволяет исследовать концентрационные профили многокомпонентных смесей вблизи всех границ раздела фаз в широких пределах варьирования плотности и температуры системы.
КИНЕТИЧЕСКИЕ ХАРАКТЕРИСТИКИ
Влияние СКФ на концентрационные зависимости скорости реакции. Эффективная энергия активации бимолекулярной реакции $E_{{{\text{AB}}}}^{{{\text{эф}}}}$ определяется через соотношение (4) как [23]
(9)
${{W}_{{12}}} = {{w}_{{{\text{AB}}}}}{\text{/}}(k_{{{\text{AB}}}}^{0}{{\theta }_{{\text{A}}}}{{\theta }_{{\text{B}}}}) = \exp [--\beta E_{{{\text{AB}}}}^{{{\text{эф}}}}],$Если СКФ выступает в роли растворителя, то данное влияние можно оценить по роли латеральных взаимодействий в изменении эффективной энергии активации химической реакции $E_{{{\text{AB}}}}^{{{\text{эф}}}}$ при варьировании температуры и давления системы. В случае очень большой величины EAB присутствие СКФ практически не влияет на скорость реакции, если она остается в кинетическом режиме. Однако по мере увеличения давления в реакционной системе за счет добавления инертных молекул СКФ можно перейти из кинетического режима бимолекулярной реакции в диффузионный.
В работе [23] был проведен подробный анализ вариантов соотношений между энергиями активации элементарных стадий химических превращений и транспортных коэффициентов диффузии и вязкости, вкладов межчастичных взаимодействий и брутто-плотностью системы при разных температурах для СКФ растворителя. Использование теории неидеальных реакционных систем позволяет разделить вклады от температуры и давления в СК-процессах: если преобладает вклад температуры, то отличия между $E_{{ij}}^{{{\text{эф}}}}$ и Eij малы (и можно использовать уравнения (1)). В противном случае вклад от изменения давления становится существенным, и отличия между $E_{{ij}}^{{{\text{эф}}}}$ и Eij определяют область по плотности СКФ, в которой сохраняются свойства идеальных систем.
Такая постановка отделяет химические аспекты реакционных систем от кооперативных свойств многокомпонентных смесей при высоких давлениях. Неслучайно для молекул воды превышение температуры на ~10 К выше критической температуры приводит к резкому уменьшению ее плотности, и СКФ представляет собой плотный пар вплоть до очень высоких давлений (на порядок больше критического давления Рс), которые не обсуждаются в практических приложениях [69].
На рис. 6 приведена зависимости эффективной энергии активации реакции для случая, когда СКФ является реагентом. Рассматривается влияние СКФ-компонента С на скорости бимолекулярной реакции А + С в трехкомпонентной системе А–В–С при различном содержании компонента С. В отношении $E_{{{\text{AC}}}}^{{{\text{эф}}}}$/EAC величина EAC – энергия активации реакции в идеальной системе, а $E_{{{\text{AC}}}}^{{{\text{эф}}}}$ – эффективная энергия активации той же стадии в неидеальных СК-условиях. По оси абсцисс отложено полное содержание вещества в системе θ = θA + θB + θС (здесь θA = θB). Относительный вклад латеральных взаимодействий в этом случае меньше, чем для бимолекулярной реакции типа А + В (для которой такое отношение достигает 4–5), однако концентрация СКФ входит в виде сомножителя в уравнение для скорости реакции в идеальной системе, поэтому в целом роль СК-компонента в брутто-процессе возрастает. С ростом энергии активации EAC вклад от латеральных взаимодействий уменьшается (кривые 3 и 6).
Рис. 6.
Зависимости $E_{{{\text{AC}}}}^{{{\text{эф}}}}$/EАС от степени заполнения θ при значениях εСС = 0.38 (энергии в ккал/моль), εАА = 4εСС, εВВ = 2.4εСС для Т = 1.1 Тс, Е12 = 3 (1), 10 (2), 20 (3) и Т = 2.5 Тс, Е12 = 3 (4), 10 (5), 20 (6). Кривые относятся к концентрации реагентов хА = хВ = = 0.01 мол. доли.
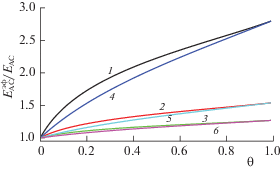
При повышении плотности инертного СКФ (θ → 1) всегда можно достигнуть такого состояния, когда реакция может перейти из области с лимитирующим химическим превращением в область, когда лимитирует транспорт реагентов, особенно при малых концентрациях реагентов. Эта проблема отсутствует, если СКФ является одним из реагентов стадии: в избытке СКФ бимолекулярная реакция переходит в квазимономолекулярную. В этом случае главным становится вопрос о химических процессах, реализуемых при высоких температурах.
В целом, при анализе роли СК-условий необходимо учитывать не только увеличение скорости реакции при повышении температуры и уменьшение скорости реакции и коэффициента самодиффузии (+ увеличение вязкости) при повышении давления, но и возможные изменения с температурой и давлением механизма процесса и состояния других материалов. Для реализации СК-процессов важен поиск оптимальных условий между скоростями реакций и подвижностью реагентов. Простое увеличение одного из параметров состояний (температуры или давления) может привести к ухудшению условий реализации элементарных стадий. При переходе к конкретным механизмам брутто-процессов теория неидеальных реакционных систем дает возможность связать кинетические характеристики, наблюдаемые в нормальных условиях при атмосферном давлении, с аналогичными характеристиками для СК-условий.
Диссипативные коэффициенты. Реальные СКФ-процессы реализуются в технологических аппаратах, которые включают в себя не только изменения скоростей конкретных реакций, но и общие теплофизические балансы, которые основаны на анализе потоков импульса, энергии и массы. Эффективный парный потенциал (8) использовался также и для расчета диссипативных коэффициентов. С этой целью были рассмотрены возможности описания диссипативных коэффициентов: вязкости и теплопроводности чистых веществ, а также коэффициенты самодиффузии, взаимной диффузии и вязкости для бинарных смесей. Как и выше, расчеты проводились как для свойств индивидуальных веществ, так и для их обобщенных диаграмм, которые отражают совокупные свойства множества родственных веществ, подчиняющихся закону соответственных состояний. Формально данному закону подчиняются многие вещества, в которых отсутствуют специфические взаимодействия, приводящие в формированию более прочных связей. Для индивидуальных веществ такого рода закономерности гарантируют точность порядка 3–5%.
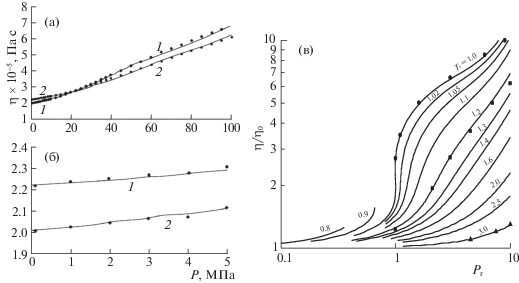
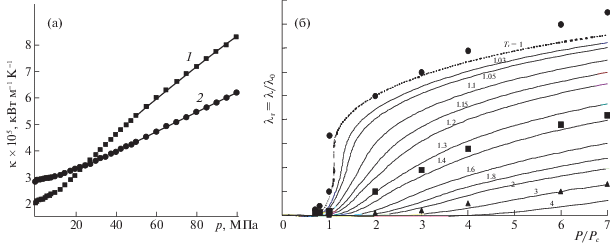
На рис. 7 показаны коэффициенты сдвиговой вязкости, которые теория описывает с точностью до ~5%.
Рис. 7.
а – Зависимости сдвиговой вязкости (η ) О2 от давления P при различных температурах газа: 1 – 289, 2 – 328 K, точки – эксперимент [70]; б – аналогичные зависимости для малых плотностей; в – обобщенная диаграмма приведенной сдвиговой вязкости η/η0 в зависимости от приведенного давления Pr при различных значениях приведенной температуры Tr, точки – эксперимент [43, 57].
Важным технологическим коэффициентом является коэффициент теплопроводности. На рис. 8а приведены эти коэффициенты для атомов аргона при Т = 273 и 400 К в объемной фазе, а на рис. 8б показаны обобщенные зависимости коэффициента теплопроводности по данным [57]. Расчет основан на использовании двух каналов механизма тепла (диффузионный – за счет движения атомов аргона, а также через механизм столкновения, впервые введенного в теории Энскога для разреженных газов [71]).
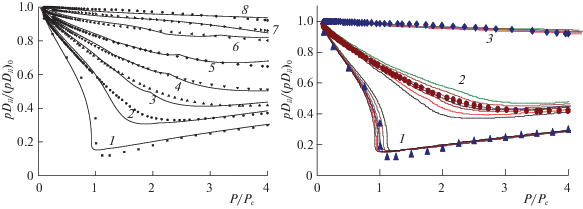
Для многокомпонентных смесей необходимы коэффициенты самодиффузии и взаимной диффузии, они приведены на рис. 9. Коэффициент самодиффузии характеризует движение метки в однокомпонентных веществах при условии равновесного распределения молекул во всей рассматриваемой области. Коэффициент взаимной диффузии характеризует неравновесие по составу смеси. Экспериментальные данные группируются по средневзвешенным корреляциям, как описано в работе [57]. Это позволяет давать оценки коэффициентов само- и взаимной диффузии для широкого диапазона давлений и температур. На рис. 9б эти данные относятся к индивидуальным газам и для смеси атомов аргона с молекулами азота.
Рис. 9.
а – Обобщенные диаграммы коэффициента самодиффузии для диапазона давлений P/Pc < 4 (линии – расчет [55], точки – эксперимент [57]) при значениях τ = 1 (1), 1.1 (2), 1.2 (3), 1.3 (4), 1.4 (5), 1.6 (6), 2 (7) и 3 (8); б – обобщенные диаграммы коэффициентов взаимной диффузии смеси Ar–N2 для ${{x}_{{{{{\text{N}}}_{{\text{2}}}}}}}$ = 0, 0.2, 0.4, 0.5, 0.6, 0.8, 1.0 мол. доли при τ = 1 (1), 1.3 (2) и 3 (3) в диапазоне давлений P/Pc < 4; рассчитанные кривые в пучке для каждого значения τ расположены снизу вверх в порядке возрастания концентрации азота [55]; точки – пересчет экспериментальных данных по правилам [57].
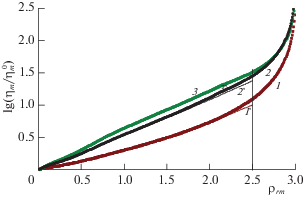
Один из наиболее популярных методов расчета вязкости смесей неполярных плотных газов был разработан Дином и Стилом (цитируется по [57]). На основании обработки большого количества экспериментальных данных по бинарным смесям легких углеводородов, углеводородов с инертными газами и компонентами воздуха была получена универсальная формула [57], которая представлена на рис. 10 тонкими линиями, отражающими эксперимент, (толстые – расчет МРГ [55]). Сопоставлены зависимости ηm/$\eta _{m}^{0}$ (нормировка на свойства разреженного газа) от приведенной плотности ρrm, полученные для бинарной смеси Ar–N2 для трех значений τ = 0.75, 1 и 3 по этой эмпирической формуле и по МРГ. Приведенные температуры τ относятся к критическим значениям для соответствующих составов смеси. При построении этих зависимостей использовался логарифмический масштаб, в котором зависимости близки к линейным. В окрестности приведенной плотности ρrm = 2.4 теоретические кривые резко уходят вверх, отражая резкое повышение вязкости в жидкой фазе.
БОЛЕЕ СЛОЖНЫЕ МОДЕЛИ
В данном разделе дан краткий перечень примеров развития теории на базе МРГ, которая отражает ключевые вопросы моделирования кинетики в СК-условиях. К ним относятся вопросы по 1) уточнению потенциальных функций, 2) статистическому описанию распределения молекул, 3) о природе критической области парожидкостных систем и 4) о переходе на общие модели для описания трехагрегатных состояний и о кинетических моделях для их эволюции, включая самосогласованное описание элементарных стадий и локального равновесия в сложных СКФ-системах.
Потенциальные функции. Один из способов улучшения описания потенциальных функций – отказ от сферически симметричных функций типа (8) и использование атом-атомных потенциальных функций для больших молекул, в том числе и учет их размера и формы. Это старый вопрос для решеточных моделей [19–22] (см. также [72–74]), который традиционно применяется для учета взаимодействия ближайших соседей, в виде взаимодействия ближайших контактов. Данный вопрос был рассмотрен в работе [75], чтобы учесть отличия размеров молекул, “твердые сферы” которых занимают нецелое число ячеек. Так, например, молекула СО2 – один из наиболее частых СКФ, имеет отношение длинной частицы молекул к ее сечению, равное 1.38. На рис. 11 показаны кривые для фактора сжимаемости χ = = PV/(PV)s, где (PV)s относится к давлению и удельному объему, взятым при нормальных условиях для молекул СО2, полученным с учетом предложенной модификации потенциала.
Рис. 11.
Сравнение экспериментального [76] (точки) и рассчитанного (линии) фактора сжимаемости для молекул CO2; расчет при Т = 273 (1, 2, 4) и 373 К (3).
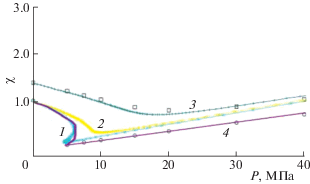
Кривые 1–3 получены при использовании одноузельной модели, кривая 4 – при использовании аппроксимационной модели для твердой сердцевины. (Кривая 2 относится к модельному расчету с сильным тройным отталкиванием соседей.) Введенная модификация лучше отражает эксперимент [76]. Кривые 1 и 4 отражают расслаивание молекул СО2 при температуре 273 К – ниже критической температуры 304 К. Следует отметить, что для кривых расслаивания парожидкостной системы оба варианта дают примерно близкие результаты, поэтому выбор модели по кривой расслаивания не является столь же чувствительным.
Данная модель взаимодействия между продолговатыми молекулами учитывает несферичность твердой сердцевины молекулы и зависимость энергии контактных взаимодействий для их разных взаимных ориентаций. В этом отношении статистическое описание ориентационных состояний молекул сохраняет пространственную дискретизацию как для молекул с кратным отношением линейных сторон молекул, но общий баланс заполненных узлов и, самое главное, влияние межчастичных потенциалов через функцию неидеальности отражает реальные размеры молекул. Сформулированные подходы к рассмотрению нецелых отношений линейных размеров молекул могут быть использованы для широкого круга веществ. Такие уточнения позволяют перекрыть весь рабочий диапазон по температурам практически для многих других сверхкритических флюидов. Пренебрежение несферичностью молекулы может приводить к изменению значений коэффициентов самодиффузии и других транспортных характеристик.
В случае поверхностных процессов и внутри пористых материалов требуется дополнительная апробация новой техники учета несоразмерности молекул, так как влияние внешнего потенциала стенки усиливает роль асимметрии твердой сферы молекулы. Однако следует отметить, что такой подход годится для описания ближнего порядка в отсутствие строгой упорядоченности молекул. Наличие дальнего порядка может быть более чувствительно к вариациям длины молекулы.
Другим активно используемым СКФ является вода. В работах [77, 78] был рассмотрен вариант потенциала, объединяющего парный ЛД- и дипольный потенциалы. Данная модель обобщает ранее проведенные расчеты в приближении среднего поля [79, 80] на случай учета эффектов корреляции в КХП. Недостаточность использования одного электростатического взаимодействия также давно известна [5–9]. Учёт же обоих эффектов позволяет с количественной точностью воспроизводить ряд термодинамических свойств воды [81, 82]. Такую же потенциальную возможность можно предположить и для других дипольных жидкостей, так как модель отражает атомистическую природу вещества без привлечения макроскопических характеристик типа диэлектрической проницаемости. В частности, модель показала, что кривую расслаивания пар–жидкость для водных систем она описывает практически с той же точностью, что и кривую расслаивания для аргона (отличия между двумя системами обусловлены только самим потенциалом). С другой стороны, та же модель [78] позволяет описать [83] экспериментальные данные [82] по самодиффузии молекул воды.
Распределения молекул. Вопросами статистики в кинетике достаточно часто пренебрегают. Самым известным примером такого некорректного использования статистики является использование в ТАСР представлений о коэффициенте активности АК – уравнения (2) и (3). Анализ применимости ТАСР к конденсированным фазам в виде уравнений (3) показал [64, 83–86], что в них нарушено основное условие ТАСР о наличии равновесия между АК и окружающими молекулами. Так как времена релаксации окружения соседей много больше, чем время формирования АК, использование уравнений (3) вместо уравнений (4) может приводить к заметным отличиям в величинах скоростей, особенно при малых и больших плотностях. Аналогичное замечание относится и к скоростям мономолекулярных стадий. Уравнения, связывающие между собой константы скоростей элементарных химических стадий для экспериментов, проведенных при атмосферном давлении, и для сверхкритических условий, которые заменяют традиционные уравнения ТАСР (3) с использованием коэффициента активности для активированного комплекса, обсуждаются в работе [85].
Теория на основе МРГ обеспечивает получение самосогласованного описания равновесного состояния реакционной смеси и скоростей элементарных стадий: его суть состоит в том, что приравнивая скорости противоположно направленных процессов любой стадии, получают уравнение равновесного распределения компонентов. Этим свойством обладают только приближения, учитывающие ближние корреляции типа КХП. Все приближения, в которых допускается некоррелированное распределение частиц не обеспечивают самосогласованности описания скоростей стадий и равновесия (подробнее см. [10, 11, 87, 88]).
Важным фактором уточнения статистики описания распределений компонентов смесей в растворах является явный учет степеней свободы всех компонентов. Все традиционные принципы построения скоростей реакций в рамках ТАСР (см. например, [6, 12, 17, 18, 89]) исключают в плотных фазах описание межмолекулярных колебаний, неявно или явно предполагая, что реагенты имеют внутренние статсуммы, отделимые от их трансляций и вращений. Однако в работе [90] было показано, что учет зацеплений межмолекулярных колебаний и внутримолекулярных колебаний и трансляций компонентов резко усложняет структуру выражений для их равновесного распределения.
О природе критической области. Обсуждая вопросы статистики, следует отметить, что хотя выяснение природы критического состояния, относительно которого в большинстве случаев считается достаточным ограничиться скейлинговым приближением [58–60], продолжается, она остается в центре внимания макроскопических подходов. Это касается уточнения методов описания акустических измерений [91] и термодинамических трактовок [92]. В первом случае авторы включили учет коллективных колебательных движений молекул в рамках макроскопической модели Дебая, а во втором случае упор сделан на анализе роли изохорной теплоемкости как ключевого термодинамического свойства вещества в исследовании критических и СК-явлений. Такое рассмотрение выявляет физическую природу асимметрии кривой сосуществования жидкость– газ вблизи критической точки и оценивает вклад химического потенциала в расходимость двухфазной изохорной теплоемкости в критической точке. В обеих работах [91, 92] уделено внимание СК-фазовым переходам и определению линии Widom, которая отделяет жидкоподобную область от газоподобной в СК-состояниях, т.е. определяет границы сосуществования жидко- и газоподобных фаз в СКФ.
Вместе с тем, эти макроскопические подходы мало внимания уделяют эффектам ближнего порядка, которые собственно и определяют кинетику элементарных стадий и механизмы их протекания. Реакция реализуется в малом элементарном объеме, содержащем реагенты, и здесь в первую очередь важны корреляции ближайших соседей. Это тот аспект, которому уделяется главное внимание в МРГ. В теории скейлинга [58–60] центральная величина теории Ткрит считается особой точкой, и она берется из эксперимента. Расчет величины Ткрит тесно связан с эффектами корреляции. В то же время ее значения зависят от локальных корреляций и точности их описания. Для химической кинетики и процессов фазообразования важны локальные эффекты, влияющие на локальные активационные энергии/барьеры. Для согласования описания эффектов ближнего порядка и крупномасштабных флуктуаций была введена калибровочная функция [61–64], которая играет роль поправочной функции и позволяет получить квазиточные значения термодинамических функций и соответственно скоростей элементарных стадий.
Следует также напомнить, что для успешной реализации химических превращений в СК-условиях необходимо смещаться по температуре вверх от критической точки фазовых переходов первого рода как в твердых, так и в жидких фазах. Иначе вблизи критической точки замедляются процессы выравнивания плотности (затрудняются транспортные, т.е. диффузионные стадии), и это затрудняет течение химических многостадийных процессов [93–96].
Кинетические уравнения. Усложнение математических моделей происходит при увеличении числа стадий в брутто-процессе, при наличии влияния внешних полей, при наличии в системе нескольких фаз и наличии разномасштабных времен релаксаций в неравновесности состояний реагентов в разных фазах. Во всех этих случаях необходимо использовать для моделирования кинетические уравнения.
Эти уравнения должны быть построены на основе выражений для элементарных стадий с учетом неидеальности реакционной системы [10, 11], а также новых разработок, указанных выше. Так, все уравнения состояния и построения для диссипативных коэффициентов, указанные выше, были обобщены на сложные пористые материалы, в которых реализуются как фазовые, так и СК-состояния [64]. Стенки пор формируют внешние поля, внутри которых распределяются пар и жидкость, а также внутри которых возможно протекание химических/каталитических реакций. На базе этих походов была сформулирована микроскопическая гидродинамика [64]. Точно также методы [86], разработанные для процессов формирования новых фаз, естественным образом отражают процессы, реализуемые в СК-условиях. Учет различий в размерах молекул, обсуждаемый выше, был включен в кинетические уравнения [97], а наличие заряженных частиц в системе, и в частности электрона, было отражено в уравнениях [98, 99].
Общая тенденция разработок кинетических уравнений в МРГ позволила сформулировать общие подходы к трехагрегатным системам [86, 100] и с единых позиций рассматривать самые разнообразные процессы. К ним относятся часто встречающиеся в СКФ-процессы изменения фазового состояния материалов под влиянием как самого СКФ, так и при сопутствующих химических реакциях. В качестве примера отметим работу по разработке основ молекулярной модели набухания полимерных матриц под влиянием СК- флюида СО2 [97, 101]. В данной работе изложен метод построения кинетических уравнений процессов набухания полимерной матрицы при сорбции низкомолекулярных молекул на базе МРГ. Несмотря на актуальность проблемы, до настоящего времени не существует полной молекулярной теории набухания полимеров в СКФ.
Работы [23, 68] указывают на возможности СКФ достаточно сильно влиять на состояния адсорбентов и катализаторов (разрыхлять материалы и стабилизировать химически активные частицы), что нашло свое качественное подтверждение при изучении совместного влияния СКФ-СО2 и озона на коксоподобные отложения на поверхности биметаллических катализаторов риформинга Pt-Re/γ-Al2O3102.
ЗАКЛЮЧЕНИЕ
Давно отмечено [103], что по точности расчета всех термодинамических характеристик, исключая структурные свойства, МРГ не уступает методам Монте-Карло, молекулярной динамики и интегральным уравнениям. Дать подробный обзор работ в многочисленных областях приложений МРГ не представляется возможным. Последующие разработки [10, 11] статистической техники МРГ для сильно неоднородных систем (в задачах адсорбции и абсорбции) позволили усилить данную технику, а переход на учет внутренних движений молекул внутри ячеек МРГ [27, 28, 33] позволил перейти к единому описанию трехагрегатных систем [86, 101]. Эти шаги, естественно, приводят к возможности работы с более сложными многочастичными потенциалами, в том числе и с квантово-химическими расчетами межмолекулярных взаимодействий [10, 11, 104, 105], а также и с кулоновским потенциалом [99, 106]. Статистическое обоснование потенциальных кривых типа (8) устраняет подгоночный характер энергетических параметров и позволяет оперировать с межмолекулярными потенциалами взаимодействия, как и во всех других методах молекулярного уровня. Решение обратных задач в МРГ реализуется значительно быстрее, стабильней и точнее, чем в других методах, что позволяет искать потенциальные функции из соответствующих экспериментов. Молекулярные параметры МРГ: размер ячейки, доля “исключенного” объема, и вклад колебаний ближайших связанных молекул, не требуют дополнительных параметров. Они определяются только из свойств потенциальных функций при минимизации свободной энергии по параметрам разбиения объема на ячейки.
Как и термодинамический подход [107, 108], МРГ применима к любой из фаз. Современные молекулярно-статистические подходы на основе МРГ [10, 11, 64, 86] применяются для моделирования: характера распределения компонентов гетерогенных систем и их фазовых диаграмм в широкой области изменения температур и давлений, химических реакций синтеза неорганических и органических соединений, включая инициирование и рост полимерных цепей, процессов адсорбции, катализа и роста кристаллов, стадий транспорта через разнообразные пористые материалы, мембраны и тонкие пленки, реологических свойств молекулярных и полидисперсных систем и других аспектов физико-химической механики, и т.д.
Для задач СК-процессов теория позволяет исследовать влияние плотности и температуры на все перечисленные процессы, в частности на скорости элементарных реакций, процессы адсорбции и абсорбции в пористые материалы, вязкости флюида, деформируемости материалов и т.д., по сравнению с обычными условиями при низких температурах и давлении в 1 атм.
Основы разработанных походов на базе МРГ могут быть применены для: 1) оценки характерных времен для стадии набухания полимерных матриц под воздействием СК-СО2; 2) моделирования синтеза наноразмерных частиц в СКФ объемных фазах как процесс роста новой фазы, анализа кластеризации наночастиц при восстановлении, роль температуры, другие факторы, влияющие на размер кластера; 3) анализа концентрационных эффектов за счет молекул СКФ и реагентов при разных соотношениях мольных долей, с учетом стерического фактора СК-молекул (учет их влияния на критическую температуру и на смещения констант равновесий и реакций); 4) факторов, влияющие на кинетику химических реакций и константы равновесия в пористых системах в присутствии СК-среды, особенно, специфика роли потенциала стенок для процессов роста новой фазы в порах и на поверхностях твердых тел в СК-условиях; 5) влияния перехода системы через критические точки в объемной фазе и внутри пористых тел на процессы с учетом включения молекул СКФ в химические реакции, транспорт в порах, отвод тепла, и т.д.
Разработанные математические модели обеспечивают выход на уровень моделирования равновесных состояний, скоростей реакций, транспортных характеристик и замедленных релаксационных процессов в трехагрегатных состояниях. По точности МРГ теория не уступает, а по быстродействию, не менее, чем на 2–4 порядка быстрее по сравнению с методом молекулярной динамики.
Список литературы
Галкин А.А., Лунин В.В. // Успехи химии. 2005. Т. 74. № 1. С. 24.
Savage P.E., Gopalan S., Mizan T.I. et al. // AIChE Journal. 1995. V. 41. № 7. P. 1723.
Залепугин Д.Ю., Тилькунова Н.А., Чернышова И.В., Поляков В.С. // Сверхкрит. флюиды. Теория и практика. 2006. Т. 1. № 1. С. 27.
Богдан В.И., Коклин А.Е., Казанский В.Б. // Там же. 2006. Т. 1. № 2. С. 5.
Энтелис С.Г., Тигер Р.Л. Кинетика реакций в жидкой фазе. М.: Химия, 1973. 416 с.
Мелвин-Хьюз Б.А. Равновесие и кинетика реакций в растворах. М.: Химия, 1975. 470 с.
Marcus R.A. // Ann. Rev. Phys. Chem. 1964. V. 15. P. 1.
Догонадзе Р.Р., Кузнецов А.М. // Итоги науки и техники. Кинетика и катализ. Т. 5. М.: ВИНИТИ, 1978. С. 5.
Kuznetsov A.M., Ulstrup J. Electron Transfer in Chemistry and Biology. Chichester: John Wiley & Sons, Ltd. 1999. 350 p.
Товбин Ю.К. Теория физико-химических процессов на границе газ–твердое тело. М.: Наука, 1990. 288 с.
Tovbin Yu.K. // Progress in Surface Sci. 1990. V. 34. P. 1–236.
Глесстон С., Лейдлер К., Эйринг Г. Теория абсолютных скоростей реакций. М.: Изд-во иностр. лит., 1948. 583 с. (S. Glasstone, K. Laidler, H. Eyring, The Theory of Rate Processes, Princeton Univ., New York, 1941.)
Бенсон С. Основы химической кинетики. М.: Мир, 1964. 308 с.
Еремин Е.Н. Основы химической кинетики. М.: Высшая школа, 1976. 374 с.
Темкин М.И. // Журн. физ. химии. 1938. Т. 11. № 2. С. 169.
Темкин М.И. // Там же. 1950. Т. 24. С. 1312.
Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука. 1964. 607 с.
Киперман С.Л. Основы химической кинетики в гетерогенном катализе. М.: Химия, 1979. 350 с.
Guggenheim E.A. Mixtures. Oxford: Clarendon Press, 1952. 271 p.
Пригожин И.Р. Молекулярная теория растворов. М.: Металлургия, 1990. 359 с. (Prigogine I.P. The Molecular Theory of Solutions. Interscience Publishers Inc., Amsterdam, New York, 1957).
Смирнова Н.А. Молекулярная теория растворов. Л.: Химия, 1987. 360 с.
Kruse A., Dinjus E. // J. Supercritical Fluids. 2007. V. 39. P. 362.
Рабинович А.Б., Товбин Ю.К. // Кинетика и катализ. 2011. Т. 52. № 4. С. 483.
Hill T.L. Statistical Mechanics. Principles and Selected Applications. N.Y.: McGraw-Hill Book Comp. Inc., 1956.
Товбин Ю.К. // Журн. физ. химии. 1981. Т. 55. № 2. С. 273.
Товбин Ю.К. // Кинетика и катализ. 1982. Т. 23. № 4. С. 821.
Товбин Ю.К // Журн. физ. химии. 1995. Т. 69 . № 1. С. 118.
Товбин Ю.К // Там же. 1998. Т. 72. № 5. С. 775.
Боголюбов Н.Н. Проблемы динамической теории в статистической физике. М.: Гостехиздат, 1946.
Фишер И.З. Статистическая теория жидкостей. М.: ГИФМЛ, 1961. 280 с.
Крокстон К. Физика жидкого состояния. М.: Мир, 1979. 400 с. (Croxton C.A., Liquid State Physics - A Statistical Mechanical Introduction. Cambridge Univer. Press. Cambridge. 1974.)
Мартынов Г.А. Классическая статическая физика. Теория жидкостей. Долгопрудный: Интеллект, 2011. 326 с.
Товбин Ю.К., Сенявин М.М., Жидкова Л.К. // Журн. физ. химии. 1999. Т. 73. С. 304.
Ono S. // Mem. Fac. Eng. Kyushi. Univ. 1947. V. 10. P. 190.
Федянин В.К. // Поверхностные явления в жидкостях. Л.: Изд-во ЛГУ, 1975. С. 232.
Баталин О.Ю., Товбин Ю.К., Федянин В.К. // Журн. физ. химии. 1979. Т. 53. № 12. С. 3020.
Плакида Н.М. Статистическая физика и квантовая теория поля. М.: Наука, 1973. С. 238.
Загребнов В.А., Федянин В.К. // ТМФ. 1972. Т. 10. С. 127.
Федянин В.К. Теоретические методы описания свойств растворов. Межвузовский сборник научных трудов. Иваново, 1987. С. 40.
Barker J.A. Lattice Theories of the Liquid State. Oxford: Pergamon Press, 1963.
Шахпаронов М.И. Введение в молекулярную теорию растворов. М.: ГИТТЛ, 1956. 507 с.
Морачевский А.Г., Смирнова Н.А., Пиотровская Е.М. и др. Термодинамика равновесия жидкость–пар / Под ред. А.Г. Морачевского. Л.: Химия, 1989.
Гиршфельдер Дж., Кертис Ч., Берд Р. Молекулярная теория газов и жидкостей. М.: Изд-во иностр. лит., 1961. 929 с. (Hirschfelder J.O., Curtiss C.F., Bird R.B. Molecular Theory of Gases and Liquids. N.Y.: Wiley. 1954.)
Rac Z., Collins M.F. // Phys. Rev. B. 1980. V. 20. P. 229.
Rodrigues S.P.J., Fernandes F.M.S.S. // J. Phys. Chem. 1994. V. 98. P. 3917.
Егоров Б.В., Комаров В.Н., Маркачев Ю.Е., Товбин Ю.К. // Журн. физ. химии. 2000. Т. 74. № 5. С. 882.
Товбин Ю.К., Комаров В.Н. // Там же. 2001. Т. 75. № 3. С. 562.
Комаров В.Н., Товбин Ю.К. // ТВТ. 2003. Т. 41. № 2. С. 1.
Товбин Ю.К. // Журн. физ. химии. 1987. Т. 61. С. 2711.
Товбин Ю.К., Комаров В.Н. // Там же. 2005. Т. 79. № 11. С. 2031.
Reid R.C., Sherwood T.K. The Properties of Gases and Liquids. (The Restimation and Correlation). New York, San Francis., Toronto, London, Sydny: MeGrav-Hill Boch Compay, 1966.
Levelt J. // Physica. 1960. V. 26. № 6. P. 361.
Рабинович В.А., Вассерман А.А., Недоступ В.И., Векслер Л.С. Теплофизические свойства неона, аргона, криптона и ксенона. М.: Стандартгиз, 1976.
Crain E.W., Santag R.E. // Adv. in Cryog. Eng. 1966. V. 11. P. 379.
Комаров В.Н., Рабинович А.Б., Товбин Ю.К. // Теплофизика высоких температур. 2007. Т. 45. № 4. С. 463.
Товбин Ю.К. // Журн. физ. химии. 2005. Т. 79. № 12. С. 2140.
Берд Р., Стюарт В., Лайтфут Е. Явления переноса. М.: Химия, 1974. 687 с.
Паташинский А.З., Покровский В.Л. Флуктуационная теория фазовых переходов. М.: Наука, 1975. 256 с.
Стенли Г. Фазовые переходы и критические явления. М.: Мир, 1973. 400 с. (Stanley H.E., Introduction to Phase Transitions and Critical Phenomena, Oxford: Clarendon Press. 1971.)
Ма Ш. Современная теория критических явлений. М.: Мир, 1980. (Ma Sh.-K. Modern Theory of Critical Phenomena, W.A.Benjamin, Inc., London, 1976.)
Товбин Ю.К. // Журн. физ. химии. 1998. Т. 72. № 12. С. 2254.
Товбин Ю.К., Рабинович А.Б., Вотяков Е.В. // Изв. АН. Сер. хим. 2002. № 9. С. 1531.
Tovbin Yu.K., Rabinovich A.B. // Langmuir. 2004. V. 20. № 12. P. 6041.
Товбин Ю.К. Молекулярная теория адсорбции в пористых телах. М.: Физматлит, 2012. 624 с. (Tovbin Yu.K. Moleculat Тheory of Аdsorption in Рorous Solids. Boca Raton, Fl.: CRC Press, 2017.)
Циклис Д.С. Плотные газы. М.: Химия, 1977. 165 с.
Cooper A.I. // J. Mater. Chem. 2000. V. 10. P. 207.
McHugh M.A., Krukonis V.J. Supercritical Fluid Extraction: Principles and Practice. Stoneham, 1994.
Рабинович А.Б., Товбин Ю.К. // Изв. РАН. Сер. хим. 2010. № 10. С. 1815.
Franck E.U. // Ber. Bunsen-Ges. Phys. Chem. 1984. V. 88. P. 829.
Анисимов М.А., Рабинович В.А., Сычев В.В. Термодинамика критического состояния. М.: Энергоатомиздат, 1990. 190 с.
Чепмен С., Каулинг Т. Математическая теория неоднородных газов. М.: Изд-во иностр. лит., 1960. 510 с.
DiMarzio E.A. // J. Chem. Phys. 1961. V. 35. P. 658.
Чандрасекар С. Жидкие кристаллы. М.: Mир, 1980. 344 с.
Базаров И.П., Геворкян Э.В. Статистическая теория твердых и жидких кристаллов, Москва: Изд-во МГУ, 1983. 262 с.
Товбин Ю.К. // Журн. физ. химии. 2012. Т. 86. № 4. С. 788.
Вукалович М.П., Алтунин В.В. Теплофизические свойства двуокиси углерода. М.: Атомиздат, 1965. 410 с.
Титов С.В., Товбин Ю.К. // Изв. АН. Сер. хим. 2011. № 1. С. 12.
Титов С.В., Товбин Ю.К. // Журн. физ. химии. 2011. Т. 85. № 2. С. 243.
Bell G.M. // J. Phys. C. 1972. V. 5. № 9. P. 889.
Bell G.M., Salt D.W. // J. Chem. Soc: Faraday Trans. Pt. 2. 1976. V. 72. № 1. P. 76.
Water. A Comprehensive Treatise. / Ed. By F. Franks. New York; London: Plenum, 1972. V. 1. 596 p.
Эйзенберг Д., Кауцман В. Структура и свойства воды. Л.: Гидрометеоиздат, 1975. 280 с.
Титов С.В., Товбин Ю.К. // Журн. физ. химии. 2013. Т. 87. № 2. С. 205.
Товбин Ю.К., Вотяков Е.В. // Там же. 1997. Т. 71. № 1. С. 271.
Товбин Ю.К., Титов С.В. // Сверхкритические флюиды: теория и практика. 2011. Т. 6. № 2. С. 35.
Товбин Ю.К. Малые системы и основы термодинамики. М.: Физматлит, 2018. 404 с. (Tovbin Yu.K. Small Systems and Fundamentals of Thermodynamics. Boca Raton, Fl.: CRC Press, 2019.)
Товбин Ю.К. // Журн. физ. химии. 2018. Т. 92. № 6. С. 929.
Товбин Ю.К. // Кинетика и катализ. 2019. Т. 60. № 4. С. 428.
Мелвин-Хьюз Е.А. Равновесие и кинетика реакций в растворах. М.: Химия, 1975. 470 с.
Товбин Ю.К. // Журн. физ. химии. 2019. Т. 93. № 4. С. 485.
Бражкин В.В., Ляпин А.Г., Рыжов В.Н. и др. // УФН. 2012. Т. 182. № 11. С. 1137.
Абдулагатов И.М., Скрипов П.В. // СКФ-ТП. 2020. Т. 15. № 1. С. 34.
Лифшиц И.М. // ЖЭТФ. 1939. Т. 9. С. 481.
Фишер И.З. // Проблемы многих тел и физика плазмы. М.: Наука, 1967. С. 204.
Кричевский И.Р. Фазовые равновесия при высоких давлениях. М.: Госхимиздат, 1963.
Боровский И.Б., Гуров К.П., Марчукова Ю.Э., Угасте Ю.Э. Процессы взаимной диффузии в сплавах / Под ред. К.П. Гурова. М.: Наука, 1973. 360 с.
Товбин Ю.К. // Хим. физика. 2011. Т. 30. № 4. С. 27.
Товбин Ю.К. // Журн. физ. химии. 2011. Т. 85. № 2. С. 289.
Товбин Ю.К. // Хим. физика. 2012. Т. 31. № 12. С. 30.
Товбин Ю.К. // Журн. физ. химии. 2017. Т. 91. № 3. С. 381.
Николаев А.Ю., Лазутин А.А., Товбин Ю.К. // Тр. Всероссийской конф. “Физико-химические аспекты технологий наноматериалов, их свойства и применение”. М., НИФХИ им. Л.Я. Карпова, 2009. Обнинск: МАН “Интеллект будущего”, 2011. С. 121–135.
Гайдамака С.Н., Тимофеев В.В., Гурьев Ю.В. и др. // Сверхкритические флюиды: Теория и практика. 2010. Т. 5. № 3. С. 76.
Barker I.A., Henderson D. // Rev. Mod. Phys. 1976. V. 46. № 4. P. 587.
Товбин Ю.К. // Журн. физ. химии. 2006. Т. 80. № 10. С. 1753.
Товбин Ю.К. // Там же. 1987. Т. 61. С. 2711.
Товбин Ю.К. // Там же. 1996. Т. 70. N 10. С. 1783
Гиббс Дж.В. Избранные труды. Термодинамика. М.: Наука. 1982. С. 9.
Пригожин И., Дефэй Р. Химическая термодинамика. Новосибирск: Наука, 1966, 510 с. (Prigogine I., Defay R., Chemical Thermodynamics. London: Longmans Green and Co. 1954.)
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии