

Журнал физической химии, 2021, T. 95, № 6, стр. 875-883
Особенности механизма коррозии низкоуглеродистых сталей в растворах кислот, содержащих соли Fe(III)
Я. Г. Авдеев a, *, Т. Э. Андреева a
a Российская академия наук, Институт физической химии и электрохимии им. А.Н. Фрумкина
119071 Москва, Россия
* E-mail: avdeevavdeev@mail.ru
Поступила в редакцию 01.07.2020
После доработки 01.07.2020
Принята к публикации 10.11.2020
Аннотация
Коррозия низкоуглеродистой стали в 2.0 M H2SO4 и 2.0 M H3PO4, содержащих соли Fe(III), изучалась по массопотере образцов металла, а также с использованием вольтамперометрических измерений на вращающемся дисковом электроде. Установлено, что коррозия стали в растворах минеральных кислот, содержащих соли Fe(III), включает реакцию анодной ионизации металлического железа, протекающую в кинетической области. Катодная реакция объединяет параллельно и независимо реализуемые выделение водорода, протекающее с кинетическим контролем, и восстановление катионов Fe(III) до Fe(II), характеризующееся диффузионным контролем. Потенциометрия и циклическая вольтамперометрия платинового электрода в растворах кислот, содержащих соли Fe(III), позволили установить, что катионы Fe(III) в этих средах связаны в комплексы с анионами кислотных остатков. Чем более прочные комплексы образуют катионы Fe(III), тем ниже их окислительная способность и коэффициент диффузии. Снижение коэффициента диффузии катионов Fe(III) в растворах минеральных кислот влияет на скорость стадии их восстановления на стали, протекающей с диффузионным контролем. Введение FePO4 в раствор H3PO4 слабее ускоряет коррозию низкоуглеродистой стали, чем эквимолярная добавка Fe2(SO4)3 в растворе H2SO4. Наблюдаемый эффект является результатом более низкого коэффициента диффузии катионов Fe(III) в растворе H3PO4, в сравнении с раствором H2SO4.
Коррозия низкоуглеродистых сталей в растворах минеральных кислот (так называемых “не окислителей”) в общем виде описывается суммарной реакцией:
которая является результатом протекания четырех парциальных реакций, а именно [1]:реакции анодного растворения железа
катодного осаждения железа
катодного выделения водорода
и его анодной ионизации
Протекание коррозии является результатом преимущественной реализации реакций (2) и (4).Существующие взгляды на катодное выделение водорода кратко рассмотрены в [1]. Эта реакция состоит из нескольких последовательных стадий – доставка H+ из объема кислоты к поверхности металла (${{({{{\text{H}}}^{ + }})}_{*}}$ – ион водорода, находящийся на самом близком расстоянии от поверхности металла)
реакции переноса заряда (реакция Фольмера)
За ней следует стадия химической (реакция Тафеля)
(8)
${{{\text{H}}}_{{{\text{ad}}}}} + {{{\text{H}}}_{{{\text{ad}}}}} \to {{({{{\text{Н}}}_{2}})}_{{{\text{ad}}}}}$(9)
${{{\text{H}}}_{{{\text{ad}}}}} + {{({{{\text{H}}}^{ + }})}_{*}} + {\text{e}} \to {{({{{\text{Н}}}_{2}})}_{{{\text{ad}}}}}.$Совокупность стадий (7) и (8) называется механизмом Фольмера–Тафеля, а (7) и (9) механизмом Фольмера–Гейровского. Существует мнение [2], что на поверхности сталей при выделении водорода реализуется оба механизма.
Столь же неоднозначен механизм анодного растворения железа [1]. Хойслер предполагает, что в ходе реакции атомов Fe кристаллической решетки с адсорбированными OH–-ионами образуется соединение FeOHad:
(10)
${\text{Fe}} + {\text{OH}}_{{{\text{ad}}}}^{ - } \to {\text{FeO}}{{{\text{H}}}_{{{\text{ad}}}}} + {\text{e}}.$Предполагается, что реакция перехода ионов Fe(II) через двойной слой катализируется этим соединением:
(11)
$\begin{gathered} {\text{Fe}} + {\text{OH}}_{{{\text{ad}}}}^{ - } + {\text{FeO}}{{{\text{H}}}_{{{\text{ad}}}}} \to \\ \to {{{\text{(FeO}}{{{\text{H}}}^{ + }})}_{*}} + {\text{FeO}}{{{\text{H}}}_{{{\text{ad}}}}} + 2{\text{e}}. \\ \end{gathered} $В кислом растворе соединений FeOH+ медленно распадается:
(12)
${{({\text{FeO}}{{{\text{H}}}^{ + }})}_{*}} + {{({{{\text{H}}}^{ + }})}_{*}} \to {{({\text{F}}{{{\text{e}}}^{{2 + }}})}_{*}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$В противоположность этому Бокрис [3] рассматривает FeOH как промежуточный продукт при стадийном протекании реакции:
(15)
${\text{FeO}}{{{\text{H}}}^{ + }} \to {\text{F}}{{{\text{e}}}^{{2 + }}} + {\text{O}}{{{\text{H}}}^{--}}.$Более сложные механизмы анодной реакции железа допускают участие в ней не только OH–, но и анионов, образующихся при диссоциации кислоты [4, 5].
Промышленная эксплуатация растворов кислот часто сопряжена с накоплением в них солей Fe(III) [6], что должно существенно повышать их агрессивность в отношении низкоуглеродистых сталей. Кроме этого, в таких средах резко снижаются возможности ингибиторной защиты сталей [7–9]. В результате присутствия в растворах кислот сильного окислителя – соли Fe(III) в системе дополнительно реализуется процесс
включающий катодную
и анодную (2) реакции. К сожалению, в доступной нам литературе отсутствует детальная информация о механизме коррозии сталей в таких средах, что ограничивает разработку подходов к защите металлов в них.В связи с вышесказанным представляется важным установить особенности механизма коррозии низкоуглеродистых сталей в растворах минеральных кислот, содержащих соли Fe(III). В качестве коррозионной среды нами была выбрана имеющая широкое промышленное применение H2SO4 и во многом близкая к ней по свойствам H3PO4. Важную информацию необходимую для понимания деталей механизма коррозии сталей в кислых растворах, содержащих соли Fe(III), позволят дать потенциометрические и вольтамперометрические исследования этих сред в ходе которых возможно дать количественную оценку их окислительной способности и подвижности в них катионов Fe(III) [10, 11].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для приготовления растворов использовали H2SO4 (“х.ч.”), H3PO4 (“х.ч.”) и дистиллированную воду. Растворы H3PO4, содержащие фосфаты Fe(III), получали реакцией Fe(OH)3, осажденного NaOH (“х.ч.”) из раствора FeCl3, с избытком H3PO4. Для приготовления раствора хлорида Fe(III) использовали FeCl3 ⋅ 6H2O (“ч.”). Растворы H2SO4, содержащие сульфаты Fe, готовили из FeSO4 ⋅ 7H2O (“х.ч.”) и Fe2(SO4)3 ⋅ 9H2O (“х.ч.”). Из-за технических сложностей получения растворов H3PO4, содержащих фосфаты Fe(II), пригодных для потенциометрических исследований их заменяли растворами, содержащими эквивалентную концентрацию FeSO4. При таком подходе неучтенное содержание ${\text{SO}}_{4}^{{2 - }}$ и ${\text{PO}}_{4}^{{3 - }}$ максимально достигало по 2.4% от общей концентрации анионов в растворе, что позволило нам пренебречь этим фактом в дальнейших обсуждениях.
Скорость коррозии стали 08ПС (состав, в мас. %: С – 0.08, Mn – 0.5, Si – 0.11, P – 0.035, S – 0.04, Cr – 0.1, Ni – 0.25, Cu – 0.25, As – 0.08, остальное Fe) в 2.0 М H2SO4 и 2.0 М H3PO4 при температуре t = 20°С определяли по потере массы образцов (≥3-х на точку) размером 50 мм × 20 мм × 0.5 мм из расчета 50 мл раствора кислоты на образец:
Δm – изменение массы образца, г; S – площадь образца, м2, τ – длительность коррозионных испытаний, ч. Продолжительность опытов – 2 ч. Перед опытом образцы зачищали на абразивном круге (ISO 9001, зернистость 60) и обезжиривали ацетоном.Электрохимические измерения на стали Ст 3 (состав, в мас. %: С – 0.14–0.22, P – 0.04 P, Si – 0.15–0.33, Mn – 0.40–0.65, S – 0.05, Cr – 0.3 Cr, Ni – 0.3, N – 0.008; Cu – 0.3 Cu, As – 0.08 As, остальное Fe) проводили на вращающемся дисковом электроде (n = 460 об./мин) в деаэрированных водородом 2.0 М H2SO4 и H3PO4 при t = 25°С. Потенциалы стали измеряли относительно хлоридсеребряного электрода. Стальной электрод зачищали наждачной бумагой М20 и обезжиривали ацетоном. Поляризационные кривые снимали с помощью потенциостата ЭЛ-02.061 в потенциодинамическом режиме при скорости поляризации 0.0005 В/с. Перед наложением поляризации электрод выдерживали в исследуемом растворе 30 мин для установления потенциала свободной коррозии Eкор, а затем снимали кривые анодной и катодной поляризации стали. После их снятия изучали зависимость катодного тока при Е = = ‒0.30 В от скорости вращения электрода (0, 460, 780, 1090, 1400 об./мин). За 30 мин до проведения исследований и в ходе выполнения опытов исследуемые растворы деаэрировались водородом, получаемым в электролизере из раствора NaOH.
Потенциометрические измерения в растворах кислот, содержащих Fe(III), выполняли в термостатируемой стеклянной электрохимической ячейке с вынесенным пространством для насыщенного хлоридсеребряного электрода. В качестве рабочего электрода использовалась гладкая платина (1.5 см2), электродом сравнения служил насыщенный хлоридсеребряный электрод. Разность потенциала между рабочим электродом и электродом сравнения определяли при помощи потенциостата ПИ-50.
Циклическую вольтамперометрию (ЦВА) платинового электрода в исследуемых средах проводили при помощи потенциостата ЭЛ-02.061 в термостатируемой стеклянной трехэлектродной ячейке с разделенным пространством для электродов. Рабочим электродом служила платиновая проволока (S = 15.9 мм2), электродом сравнения – хлоридсеребряный электрод, вспомогательный электрод – платиновая пластина (S = 1.5 см2). Сканирование потенциала проводили от 1.4 В до 0.0 В, а затем от 0.0 В до 1.4 В. Скорость сканирования составляла 0.10 B/c.
Потенциометрические и ЦВА-исследования выполнялись в средах предварительно в течение 30 мин и в ходе выполнения опытов деаэрированных аргоном (“х.ч.”). Перед проведением эксперимента рабочий Pt-электрод обезжиривали ацетоном, в течение 3 мин выдерживался в концентрированной HNO3, промывался дистиллированной водой. Диапазон t исследуемых растворов 20–95°С. Точность поддержания t составляла ±1°С.
Величины электродных потенциалов приводятся в статье по стандартной водородной шкале.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Добавка Fe2(SO4)3 в 2.0 M H2SO4 существенно увеличивает скорость коррозии в ней низкоуглеродистой стали (табл. 1). Введение 0.10 M Fe(III) в 2.0 M H2SO4 обеспечивает 45% приращения коррозионных потерь. В 2.0 M H3PO4 в отсутствии соли Fe(III) сталь корродирует медленнее, чем в 2.0 M H2SO4. Присутствие FePO4 в этой среде, при прочих равных условиях, обеспечивает меньшее приращение коррозионных потерь. Доля потери металла за счет присутствия 0.1 M FePO4 в 2.0 M H3PO4 составляет лишь 20%. Таким образом, природа кислоты влияет не только на скорость коррозии стали в ней, но и на ее ускорение добавками солей Fe(III). Более детальную информацию о протекании коррозии стали в растворах кислот, содержащих соли Fe(III), может дать изучение электродных реакций стали в этих средах.
Таблица 1.
Влияние катионов Fe(III) на скорость коррозии (k) и приращение коррозионных потерь (Δk) стали 08ПС в 2.0 М Н2SO4 и 2.0 M H3PO4, содержащих Fe(III), k и Δk в г/(м2ч)
| СFe(III), М | 2.0 М H2SO4 | 2.0 M H3PO4 | ||
|---|---|---|---|---|
| k | Δk | k | Δk | |
| 0 | 9.8 | 0 | 7.8 | 0 |
| 0.02 | 12 | 2.2 | 7.9 | 0.1 |
| 0.05 | 15 | 5.2 | 8.2 | 0.4 |
| 0.10 | 18 | 8.2 | 9.7 | 1.9 |
Характер поляризационных кривых (ПК) низкоуглеродистой стали в 2.0 M H2SO4 и 2.0 M H3PO4 указывает на протекание во всех случаях коррозии стали в области активного растворения (рис. 1, табл. 2). В 2.0 M H2SO4 наклон катодной поляризации стали (bс) соответствует теоретически предсказываемому значению 0.100 В, напротив анодная поляризация стали (ba) выше теоретического значения 0.040 В [12]. Увеличение наклона ba стали является результатом формирования в ходе растворения металла на его поверхности слоя шлама, наблюдаемого визуально. В 2.0 M H3PO4 наклон bс выше и составляет 0.125 В, что укладывается в параметры характерные для низкоуглеродистых сталей в растворах минеральных кислот. Формирование шлама на поверхности стали в 2.0 M H3PO4 происходит интенсивнее, что во многом определяет значение ba существенно выше 0.040 В.
Рис. 1.
Поляризационные кривые стали Ст3 в 2.0 M H2SO4 (а) и 2.0 M H3PO4 (б), содержащих Fe(III), М: 1 – 0, 2 – 0.02, 3 – 0.05, 4 – 0.10, n = 460 об./мин, t = 25°C.
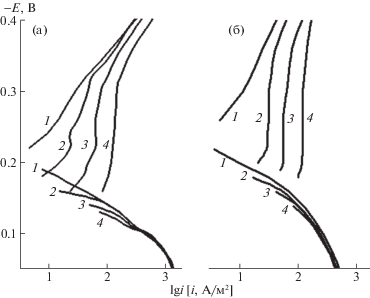
Таблица 2.
Значения потенциалов коррозии (Eкор) стали Ст3, тафелевых наклонов поляризационных кривых (bк и bа), плотности катодного и анодного токов (ic и iа) при Е = –0.30 и –0.10 В соответственно
| СFe(III), М | Екор | bс | iс | ba | ia |
|---|---|---|---|---|---|
| 2.0 M H2SO4 | |||||
| 0 | –0.20 | 0.100 | 29.2 | 0.07 | 431 |
| 0.02 | –0.17 | ilim | 46.2 | 0.07 | 477 |
| 0.05 | –0.15 | ilim | 77.9 | 0.07 | 470 |
| 0.10 | –0.14 | ilim | 142 | 0.07 | 465 |
| 2.0 M H3PO4 | |||||
| 0 | –0.23 | 0.125 | 11.5 | 0.06 | 262 |
| 0.02 | –0.19 | ilim | 31.5 | 0.06 | 227 |
| 0.05 | –0.18 | ilim | 63.1 | 0.06 | 226 |
| 0.10 | –0.17 | ilim | 119 | 0.06 | 215 |
Присутствие Fe2(SO4)3 в 2.0 M H2SO4 смещает потенциал свободной коррозии стали в область более положительных потенциалов, что является результатом растормаживания этой добавкой катодного процесса. Катионы Fe(III) практически не влияют на анодную реакцию, но наблюдается положительный порядок катодной реакции по их концентрации. Начальный участок катодных ПК характеризуется предельным током (ilim). Напротив, наклон анодной поляризации соответствует фоновой. Электрохимическое поведение стали в 2.0 M H3PO4, содержащий FePO4, имеет аналогичный качественный характер с той лишь разницей, что плотности токов катодной и анодной реакций (ic и iа) ниже.
Таким образом, присутствие в растворах кислот добавок солей Fe(III) практически не влияет на характер протекания анодной реакции. Она, как в отсутствии, так и в присутствии солей Fe(III) реализуется в соответствии с уравнением реакции (2). Напротив, характер катодных ПК указывает на участие солей Fe(III) в катодной реакции. В концентрированных растворах кислот (рН < 2) катодная реакция, соответствующая уравнению (4), протекает в области кинетического контроля [13], что хорошо согласуется с характером катодных поляризационных кривых, полученных нами. В присутствии солей Fe(III) они осложнены предельным током, что указывает на изменение механизма катодной реакции. Вероятнее всего, наблюдаемый предельный ток обусловлен диффузионными ограничениями, связанными с доставкой к поверхности стали окислителей, присутствующих в исследуемой среде. Таких окислителей два – H+ и Fe3+, но концентрация (С) первого более чем на порядок величины выше, чем второго. В таком случае предельный ток должны быть результатом диффузионных ограничений в доставке катионов Fe(III) к поверхности стали. Подтвердить это предположение можно изучив влияние потока электролита на скорость катодной реакции. Традиционным решением такой экспериментальной задачи при выполнении электрохимических исследований является применение дискового электрода, изменяя частоту вращения которого регулируют скорость потока электролита вблизи металлического электрода [13].
Сделаем допущение, что в области катодных потенциалов на стали протекают две независимых реакции восстановления H+ и Fe3+, описываемые реакциями (4) и (17). Тогда катодный ток будет складываться из двух парциальных токов, разделение которых является сложной задачей. Однако в случае, когда одна из реакций (4) протекает в кинетической области, а другая (17) в диффузионной применимо уравнение:
где ik и id – плотности кинетического и диффузионного токов. В случае ламинарного движения жидкости вблизи поверхности вращающегося металлического диска значение id прямо пропорционально корню квадратному из частоты вращения дискового электрода (n). Тогда выражение (19) принимает вид: Действительно, в обеих исследуемых средах экспериментальная зависимость ic от n1/2 имеет линейный вид (рис. 2, табл. 3). В растворах индивидуальных кислот отсутствует отклик катодного тока на изменение частоты вращения стального диска, что подтверждает кинетическую природу реакции (4). В присутствии солей Fe(III) кинетическая составляющая катодного тока такая же, как и в их отсутствии. Это свидетельствует в пользу нашего предположения о независимости реакций восстановления H+ и Fe3+. Кроме этого ясно, что реакция (4) происходит в кинетической, а реакция (17) – в диффузионной области. Необходимо констатировать, что, несмотря на более низкое содержание в растворах солей Fe(III), по сравнению с самой кислотой, даже при самой низкой частоте вращения электрода, как правило, ik < id (табл. 4).Рис. 2.
Зависимости плотности катодного тока от частоты вращения стального диска в 2.0 M H2SO4 (а) и 2.0 M H3PO4 (б), содержащих Fe(III), М: 1 – 0, 2 – 0.02, 3 – 0.05, 4 – 0.10, Е = –0.30 В, t = 25°C.
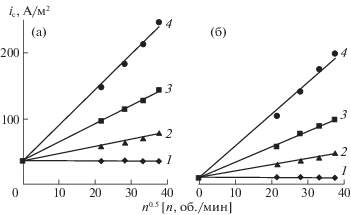
Таблица 3.
Значения постоянных ik и f в уравнении ic = ik + f n1/2 при Е = ‒0.30 В для катодной реакции стального вращающегося дискового электрода в 2.0 М H2SO4 и 2.0 M H3PO4, содержащих Fe(III), ik в А/м2, f в А мин/(м2 об.), t = 25°C
| СFe(III), М | 2.0 М H2SO4 | 2.0 M H3PO4 | ||
|---|---|---|---|---|
| ik | f | ik | f | |
| 0 | 35.3 | 0 | 10.2 | 0 |
| 0.02 | 35.3 | 1.1 | 10.2 | 0.97 |
| 0.05 | 35.3 | 2.84 | 10.2 | 2.34 |
| 0.10 | 35.3 | 5.44 | 10.2 | 4.84 |
Таблица 4.
Значения кинетического (ik) и диффузионного (id) парциальных катодных токов стального дискового электрода в 2.0 М Н2SO4 и 2.0 M H3PO4, содержащих Fe(III)
| СFe(III), М | 2.0 М H2SO4 | 2.0 M H3PO4 | ||
|---|---|---|---|---|
| ik | id | ik | id | |
| 0 | 35.3 | 0 | 10.2 | 0 |
| 0.02 | 35.3 | 23 | 10.2 | 21 |
| 0.05 | 35.3 | 62 | 10.2 | 47 |
| 0.10 | 35.3 | 112 | 10.2 | 95 |
Важно отметить, что при равном содержании солей Fe(III) в растворах кислот их ускоряющее действие на катодный процесс сильнее проявляется в случае H2SO4. Наблюдаемый эффект обусловлен природой анионов кислотных осадков, присутствующих в растворе. Предположим, что в растворах кислот катионы Fe(III) существуют не в форме гидратов, а преимущественно в виде сульфатных и фосфатных комплексов, что изменяет их свойства. На возможность формирования в растворах H2SO4 и H3PO4 катионами Fe(III) таких комплексных соединений указывают их константы нестойкости Ki (табл. 5).
Таблица 5.
Константы нестойкости (pKi) комплексов Fe(III) с некоторыми неорганическими лигандами. t = 20–30°C
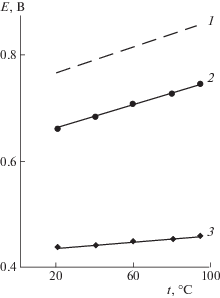
Образование комплексных соединений Fe(III) подтверждают и данные потенциометрии 2.0 М H2SO4 и 2.0 М H3PO4, содержащие эквимолярную смесь Fe(III) и Fe(II) (рис. 3). Для изучения процессов комплексообразования в этих окислительно-восстановительных системах, использован часто применяемый высокочувствительный, простой и удобный в постановке эксперимента метод потенциометрии [17, 18]. Количественной характеристикой процессов, происходящих в растворах кислот, содержащих катионы Fe(III) и Fe(II), служил реальный потенциал, который интерпретируется как потенциал окислительно-восстановительной системы. Он устанавливается в конкретном растворе при равенстве исходной концентрации окисленной и восстановленной форм потенциалопределяющих ионов без учета поправок на процессы комплексообразования, гидролиза и др. [19]. Он весьма удобен и при трактовке результатов эксперимента, связанного с комплексообразованием потенциалопределяющих ионов.
Рис. 3.
Потенциал редокс-пары Fe(III)/Fe(III): 1 – справочные данные, 2 – 2.0 M H2SO4, 2 – 2.0 M H3PO4.
Потенциал Pt-электрода в растворе, содержащем Fe(III) и Fe(II), определяется равновесной реакцией:
и рассчитывается из активных концентраций соответствующих катионов по уравнению Нернста:(22)
${{E}_{{{\text{Fe(III)/Fe(II)}}}}} = E_{{{\text{Fe(III)/Fe(II)}}}}^{{^{ \circ }}} + \frac{{RT}}{F}\ln \frac{{{{a}_{{{\text{Fe(III)}}}}}}}{{{{a}_{{{\text{Fe(II)}}}}}}}.$Более информативным методом исследования растворов кислот, содержащих Fe(III), является ЦВА. Вольтамперограммы Pt-электрода в этих средах (рис. 4, табл. 6) имеют два пика. Первый, лежащий в области более отрицательных потенциалов катодный пик, соответствует восстановлению катионов Fe(III) на Pt-электроде:
второй, анодный пик, соответствует окислению образовавшихся катионов Fe(II): Исследуемая система не является равновесной, поскольку для нее не выполняются критерии, когда разделение потенциалов анодного и катодного пиков Epa – Epc ≤ 0.057 В и соотношение токов анодного и катодного пиков Ipa/Ipc = 1. Однако на вольтамперограммах присутствуют оба пика – катодный и анодный, что позволяет трактовать ее как квазиравновесную [21].Рис. 4.
Вольтамперограммы Pt-электрода в 2.0 M H2SO4 (а) и 2.0 M H3PO4 (б), содержащих 0.10 М Fe(III), ${v}$ = 0.10 В/с.
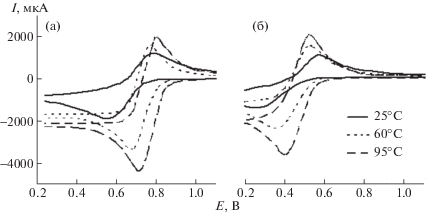
Таблица 6.
Потенциалы катодного и анодного пиков (Epc и Epa), разделение потенциалов анодного и катодного пиков (Epa – Epc), потенциалы полуволны (E1/2), токи катодного и анодного пиков (Ipc и Ipa), соотношение токов анодного и катодного пиков (Ipa/Ipc) Pt-электрода в 2.0 M H2SO4 и 2.0 M H3PO4, содержащих Fe(III)
| t, °C | Epc | Epa | Epa – Epc | E1/2 | Ipc | Ipa | Ipa/Ipc |
|---|---|---|---|---|---|---|---|
| 2.0 M H2SO4 + 0.10 M Fe(III) | |||||||
| 25 | 0.55 | 0.78 | 0.23 | 0.67 | 1.8 | 1.7 | 0.94 |
| 40 | 0.63 | 0.75 | 0.12 | 0.69 | 2.6 | 2.8 | 1.08 |
| 60 | 0.68 | 0.77 | 0.09 | 0.73 | 3.4 | 3.1 | 0.91 |
| 80 | 0.70 | 0.79 | 0.09 | 0.75 | 3.8 | 3.2 | 0.84 |
| 95 | 0.71 | 0.80 | 0.09 | 0.76 | 4.3 | 4.0 | 0.93 |
| 2.0 M H3PO4 + 0.10 M Fe(III) | |||||||
| 25 | 0.25 | 0.56 | 0.31 | 0.41 | 1.4 | 0.99 | 0.71 |
| 40 | 0.28 | 0.54 | 0.26 | 0.41 | 1.8 | 1.4 | 0.78 |
| 60 | 0.35 | 0.52 | 0.17 | 0.44 | 2.4 | 2.3 | 0.96 |
| 80 | 0.38 | 0.53 | 0.15 | 0.46 | 3.1 | 2.9 | 0.94 |
| 95 | 0.39 | 0.52 | 0.13 | 0.46 | 3.7 | 3.3 | 0.89 |
Окислительная способность катионов Fe(III) ниже в фосфорнокислой среде, в сравнении с растворами H2SO4. Об этом свидетельствуют значения потенциалов полуволны:
часто трактуемый как окислительно-восстановительный потенциал системы. При этом в исследуемых средах при прочих равных условиях значения E1/2 близки к $E_{{{\text{Fe(III)/Fe(II)}}}}^{{^{ \circ }}},$ определенным потенциометрическим методом. Также в 2.0 М H3PO4 более низкие значения Ipc и Ipa, в сравнении с сернокислой средой. Увеличение t вызывает рост Ipc и Ipa.Значение максимумов токов вольтамперных кривых характеризуется уравнением Рэндлса–Шевчика:
где z – число электронов, участвующих в редокс-процессе (z = 1 для редокс-пары Fe(III)/Fe(II)); F – число Фарадея (96500 Кл/моль); S – площадь поверхности электрода (м2); С – концентрация электрохимически активного вещества (моль/м3); D – коэффициент диффузии (м2/с); $v$ – скорость развертки потенциала (В/с); R – универсальная газовая постоянная, T – абсолютная температура. P – параметр, являющийся функцией $zFv\tau {\text{/}}RT$, где τ – время.Уравнение Рэндлса–Шевчика описывает редокс-процессы, реализующиеся в равновесных системах. Однако отмечается, что его можно применять для описания квазиобратимых и даже необратимых систем [21]. Рассчитанные с применением уравнения (26) коэффициенты диффузии катионов Fe(III) (DFe(III)) в фосфорной кислоте существенно ниже, чем в серной, что является результатом связывания их с фосфат-анионами в более прочные комплексы. Для рассматриваемых сред зависимость DFe(III) от t имеет вид, сходный с уравнением Аррениуса [13]:
где D0 – предэкспоненциальный множитель, W – энергия активации диффузионного процесса (рис. 5, табл. 7). Как и следовало ожидать, во всех случаях значения W достаточно низкие, что свойственно диффузионным процессам.Рис. 5.
Температурные зависимости коэффициентов диффузии катиона Fe(III) в 2.0 M H2SO4 (1) и 2.0 M H3PO4 (2).
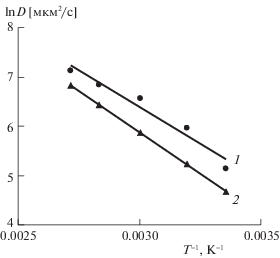
Таблица 7.
Параметры диффузии катионов Fe(III) в 2 M минеральных кислотах, вычисленные из данных ЦВА
| t, °C | 2.0 M H2SO4 | 2.0 M H3PO4 | ||||
|---|---|---|---|---|---|---|
| D × × 10–3, мкм2/с | D0 × × 10–6, мкм2/с | W, кДж/ моль | D × × 10–3, мкм2/с | D0 × × 10–6, мкм2/с | W, кДж/ моль | |
| 25 | 0.17 | 4.7 | 25 ± 1 | 0.11 | 8.9 | 28 ± 1 |
| 40 | 0.39 | 0.19 | ||||
| 60 | 0.71 | 0.35 | ||||
| 80 | 0.94 | 0.62 | ||||
| 95 | 1.25 | 0.93 | ||||
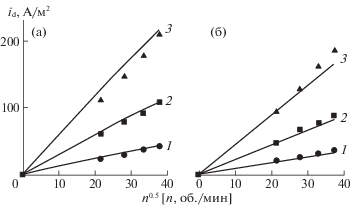
Экспериментальное определение DFe(III) в растворах кислот позволяет сравнить найденные нами опытным путем плотности тока диффузии, обусловленного восстановлением катионов Fe(III) на стальном катоде с рассчитанными теоретически для ламинарного течения жидкости [13]:
(28)
${{i}_{{\text{d}}}} = 0.62zFC{\kern 1pt} {\text{*}}{{D}^{{2/3}}}{{\eta }^{{--1/6}}}{{n}^{{1/2}}},$Рис. 6.
Зависимости плотности диффузионного тока от частоты вращения стального диска в 2.0 M H2SO4 (а) и 2.0 M H3PO4 (б), содержащих Fe(III), М: 1 – 0.02, 2 – 0.05, 3 – 0.10, Е = –0.300 В, t = 25°C. Точки – экспериментальные данные, линии – расчетная зависимость.
Коррозия сталей в растворах минеральных кислот, содержащих Fe(III), складывается из анодной реакции ионизации железа (2), а также параллельно протекающих катодных реакций – выделения водорода (4) и восстановления Fe(III) (17). Реакции (2) и (4) протекают с кинетическим контролем. Возможные механизмы реализации этих реакций обсуждались выше. Детальное установление механизма реакций (2) и (4), реализующихся в исследуемой нами системе, не входит в задачи исследования и потребовало бы серьезного дополнительного эксперимента. Более важно, что катодная реакция (17) реализуется параллельно и независимо от реакции (4). Она протекает с диффузионным контролем. Скорость катодной реакции зависит от коэффициента диффузии катионов Fe(III) в коррозионной среде, который в свою очередь определяется природой анионов кислотных остатков, присутствующих в растворе. Чем прочнее анионы кислотных остатков связывают катионы Fe(III) в комплексы, тем сильнее снижается их окислительная способность и, главное, подвижность в агрессивной среде. Факт существования катионов Fe(III) в растворах минеральных кислот в форме комплексных соединений описан в литературе [19] и подтвержден нами результатами потенциометрических исследований изучаемых систем. Идентификация присутствующих в кислотах комплексов Fe(III) является во многом сложной отдельной экспериментальной задачей. Так в работе [23] изучены химические формы существования Fe(III) в растворах H3PO4.
Таким образом, результаты коррозионных и электрохимических исследований коррозии стали в растворах минеральных кислот, содержащих соли Fe(III), находятся в полном соответствии. Стимулирующее действие солей Fe(III) на коррозию стали является результатом их катодного восстановления, протекающего с диффузионным контролем.
Таким образом, коррозия стали в растворах минеральных кислот, содержащих соли Fe(III), включает реакцию анодной ионизации металлического железа, протекающую в кинетической области. Катодная реакция объединяет параллельно и независимо реализуемые выделение водорода, протекающее с кинетическим контролем, и восстановление катионов Fe(III) до Fe(II), характеризующееся диффузионным контролем. Значения коэффициентов диффузии катионов Fe(III) в растворах минеральных кислот определяются способностью анионов кислотных остатков связывать их в комплексные соединения. Чем прочнее Fe(III) связано в комплексное соединение, тем ниже значения его окислительного потенциала и коэффициента диффузии. Снижение коэффициента диффузии катионов Fe(III) в растворах минеральных кислот влияет на скорость стадии их восстановления на стали, протекающей с диффузионным контролем. С использованием метода ЦВА определены коэффициенты диффузии катионов Fe(III) в растворах H2SO4 и H3PO4 в диапазоне t = 25–95°С. Наиболее низкие значения коэффициентов диффузии катионов Fe(III) наблюдаются в растворе H3PO4, что является следствием более прочного связывания Fe(III) в комплексы фосфат-анионами. Температурная зависимость коэффициентов диффузии катионов Fe(III) в растворах минеральных кислот от температуры имеет вид, сходный с уравнением Аррениуса. Введение FePO4 в раствор H3PO4 слабее ускоряет коррозию низкоуглеродистой стали, чем эквимолярная добавка Fe2(SO4)3 в растворе H2SO4. Наблюдаемый эффект является результатом более низкого коэффициента диффузии катионов Fe(III) в растворе H3PO4, в сравнении с раствором H2SO4.
Исследование выполнено в рамках Программы фундаментальных научных исследований государственных академий наук на 2013–2020 годы. Тема “Развитие фундаментальных научных основ защитного действия ингибиторов коррозии металлов в газовых и конденсированных средах, нанокомпозитах, лакокрасочных и конверсионных покрытиях” (№ госрегистрации АААА-А18-118121090043-0).
Список литературы
Кеше Г. Коррозия металлов. Физико-химические принципы и актуальные проблемы / Пер. с нем. под. ред. акад. Я.М. Колотыркина. М.: Металлургия, 1984. 132 с.
Антропов Л.И. Теоретическая электрохимия. М.: Высш. школа, 1965. С. 348.
Bockris J.O’M., Drazic D., Despic A.R. // Electrochim. Acta. 1961. V. 4. № 2–4. P. 325. https://doi.org/10.1016/0013-4686(61)80026-1
Florianovich G.M., Sokolova L.A., Kolotyrkin Ya.M. // Electrochim. Acta. 1967. V. 12. № 7. P. 879. https://doi.org/10.1016/0013-4686(67)80124-5
Chin R.J., Nobe K. // J. of The Electrochemical Society. 1972. V. 119. № 11. P. 1457. https://doi.org/10.1149/1.2404023
Avdeev Ya.G. // Int. J. Corros. Scale Inhib. 2019. V. 8. № 4. P. 760. https://doi.org/10.17675/2305-6894-2019-8-4-1
Авдеев Я.Г., Подобаев Н.И. // Коррозия: материалы, защита. 2004. № 12. С. 25.
Авдеев Я.Г., Белинский П.А., Кузнецов Ю.И., Зель О.О. // Коррозия: материалы, защита. 2009. № 1. С. 20.
Авдеев Я.Г., Панова А.В., Андреева Т.Э., Кузнецов Ю.И. // Коррозия: материалы, защита. 2019. № 11. С. 32. https://doi.org/10.31044/1813-7016-2019-0-11-32-40
Avdeev Ya.G., Andreeva T.E., Kuznetsov Yu.I. // Int. J. Corros. Scale Inhib. 2018. V. 7. № 3. P. 366. https://doi.org/10.17675/2305-6894-2018-7-3-7
Avdeev Ya.G., Andreeva T.E., Panova A.V., Yurasova E.N. // Int. J. Corros. Scale Inhib. 2019. V. 8. № 2. P. 411. https://doi.org/10.17675/2305-6894-2019-8-2-18
Решетников С.М. Ингибиторы кислотной коррозии металлов. Л.: Химия, 1986. 144 с.
Плесков Ю.В., Филиновский В.Ю. Вращающийся дисковый электрод. М.: Наука, 1972. 344 с.
Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1971. С. 265.
Casas J.M., Crisóstomo G., Cifuentes L. // Hydrometallurgy. 2005. V. 80. № 4. P. 254. https://doi.org/10.1016/j.hydromet.2005.07.012
Yue G., Zhao L., Olvera O.G., Asselin E. // Hydrometallurgy. 2014. V. 147–148. P. 196.https://doi.org/10.1016/j.hydromet.2014.05.008
Рахимова М.М., Нурматов Т.М., Юсупов Н.З. и др. // Журн. неорган. химии. 2013. Т. 58. № 6. С. 813.
Рахимова М.М., Юсупов Н.З., Суяров К.Дж. и др. // Журн. неорган. химии. 2013. Т. 58. № 8. С. 1090.
Захаров В.А., Сонгина О.А., Бектурова Г.Б. // Журн. аналит. химии. 1976. Т. 31. № 11. С. 2212.
Techniques of electrochemistry: Electrode Processes. V. 1 / Eds.: E. Yeager and A.J. Salkind. New York: Published by John Wiley & Sons Inc, 1972. 592 p.
Плэмбек Дж. Электрохимические методы анализа. Пер. с англ. М.: Мир, 1985. 496 с.
Краткий справочник физико-химических величин / Под ред. К.П. Мищенко и А.А. Равделя. Л.: Химия, 1967. С. 103.
Филатова Н.Л., Вендило А.Г., Санду Р.А. // Журн. неорган. химии. 2012. Т. 57. № 9. С. 1355.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии