

Журнал физической химии, 2021, T. 95, № 7, стр. 1027-1035
Термодинамика реакций комплексообразования ионов d-металлов с анионами глицина и глицилглицина в водно-органических растворителях
В. А. Исаева a, В. А. Шарнин a, К. В. Граждан a, *, К. А. Кипятков a
a Ивановский государственный химико-технологический университет
Иваново, Россия
* E-mail: grakonst@gmail.com
Поступила в редакцию 11.07.2020
После доработки 01.09.2020
Принята к публикации 14.09.2020
Аннотация
Обобщены собственные и литературные данные по термодинамическим характеристикам реакций образования комплексных соединений ионов d-металлов с глицинат- и глицилглицинат-ионами в водно-органических растворителях. Дана оценка вкладов пересольватации реагентов в изменение устойчивости глицинатных и глицилглицинатных комплексов и тепловые эффекты реакций их образования при переходе из воды в водно-органические смеси. Установлено, что в водно-органических растворах при образовании пептидных комплексов d-металлов, в отличие от аминокислотных, сольватационный вклад лиганда в изменение термодинамических параметров реакции значительно превышает различие в изменении сольватного состояния комплексного и центрального ионов.
Комплексные соединения аминокислот и пептидов с ионами d-металлов представляют интерес с точки зрения их биологической активности и находят широкое применение в фармакологии и косметологии [1]. Использование неводных и смешанных растворителей позволяет изменять растворимость комплексов, повышать эффективность трансдермального переноса биологически активных веществ [2], оптимизировать условия синтеза комплексных соединений [3, 4]. Изучение связи между термодинамическими функциями процессов комплексообразования и сольватации частиц в водно-органических средах дает возможность прогнозировать термодинамические параметры реакций, протекающих в неводных и смешанных растворителях.
Большой объем экспериментальных данных, связанных с исследованиями реакций комплексообразования ионов d-металлов с лигандами класса аминов, позволил выявить ряд закономерностей во влиянии растворителя на термодинамику этих процессов [5]. Показана применимость этих закономерностей к реакциям комплексообразования с участием заряженных лигандов, исходя из данных по константам устойчивости ацетатов и глицинатов d-металлов в водных растворах спиртов и диметилсульфоксида [6]. В настоящей работе обобщены собственные и литературные данные по устойчивости комплексных соединений d-металлов с глицинат-ионом и глицилглицинат-ионом и тепловым эффектам реакций их образования в различных водно-органических растворителях. Расширение круга рассматриваемых процессов комплексообразования, привлечение результатов термохимических исследований, сопоставление и анализ этих данных с учетом изменения термодинамических параметров пересольватации реагентов позволяют выявить особенности влияния растворителя на реакции образования комплексов с заряженными N-, O-донорными лигандами, содержащими пептидную группу.
Анион глицина (NH2CH2COO−) образует комплексные частицы с ионами двухвалентных d-металлов через атомы азота аминогруппы и кислорода карбоксилатной группы [7]. C ионом серебра(I), не склонным к хелатообразованию, более вероятно образование линейных комплексов посредством атома азота аминогруппы [8, 9]. Анион глицилглицина (NH2CH2CONHCH2COO−) способен образовывать комплексы за счет наличия трех потенциально возможных центров координации: аминогруппы, карбоксилатной группы и пептидной группы. Установлено [9, 10], что с ионами d-металлов глицилглицинат-ион образует комплексы посредством донорных атомов азота аминогруппы и кислорода пептидной группы. Процесс комплексообразования глицилглицинат-иона с ионом меди(II) имеет особенность, связанную с протеканием реакции диссоциации пептидной группы, что приводит к образованию как нормальных, так и депротонированных глицилглицинатов меди(II) [10]. При образовании депротонированных комплексов меди(II) координация смещается с пептидного кислорода на пептидный азот [10].
В настоящей работе рассматриваются изменение устойчивости и тепловые эффекты реакций образования монолигандных комплексов d-металлов с глицинат-ионом и глицилглицинат-ионом, образованных без депротонирования пептидной группы. Константы устойчивости глицинатов меди(II), никеля(II) и серебра(I) в водных растворах этанола (EtOH), изопропанола (i‑PrOH), ацетона (MeAc), диметилсульфоксида (DMSO) определены в работах [11–16], никеля(II), марганца(II), цинка(II) в водных смесях с метанолом (MeOH), диоксаном (DOX), ацетонитрилом (MeCN), диметилформамидом (DMF) – в [17], серебра(I) в смеси вода–диоксан – в [18]. Константы устойчивости глицилглицинатов никеля(II), меди(II) в водных растворах этанола, ацетона, диметилсульфоксида приведены в [19–23], кобальта(II) в растворителе вода–EtOH в [24]. Тепловые эффекты реакций образования глицинатов никеля(II) и серебра(I) в водно-этанольных и водно-диметилсульфоксидных растворах определены в работах [25–28], глицината меди(II) и глицилглицината никеля(II) в растворителе вода – DMSO – в [29, 30]. Изменение энергии Гиббса и изменение энтальпии пересольватации ионов металлов в водно-органических растворителях приводятся в работах [31–33]. Изменение энтальпии пересольватации в смесях воды с этанолом и диметилсульфоксидом глицинат-иона взято из [26, 34], глицилглицинат-иона – из [35, 36]. Изменение энергии Гиббса переноса глицинат-иона и глицилглицинат-иона из воды в водно-спиртовые смеси приведено в [37, 38], водно-диметилсульфоксидные смеси – в [39, 40]. Для глицинатов никеля(II) и серебра(I) константы устойчивости в воде и смесях воды с ацетоном и диметилсульфоксидом получены при Т = 298 К и ионной силе растворов µ = 0.3 (NaClO4), в прочих случаях константы определены при Т = 298 К и µ = 0.1 (NaClO4). Тепловые эффекты рассматриваемых реакций комплексообразования определены при Т = 298 К, для глицинатов Ag(I) и Ni(II) в растворителе вода–диметилсульфоксид при µ = = 0.3 (NaClO4), в прочих случаях при µ = 0.1 (NaClO4). Значения ΔtrGr и ΔtrHr, отнесенные к указанным условиям, при оценке сольватационных вкладов реагентов в изменение термодинамических параметров реакций комплексообразования использовали наряду со стандартными значениями ΔG° и ΔН° переноса реагентов из воды в водно-органические растворители.
Анионы глицина (G–) и глицилглицина (GG–) в водных растворах образуют с ионами переходных металлов комплексные частицы с достаточно высокой константой устойчивости (рис. 1, 2), что характерно для комплексов с аммиаком и алифатическими N-донорными лигандами [41–43 и др.]. При переходе от воды к водно-органическим смесям прирост устойчивости аминокомплексов, как правило, не превышает 1 лог. единиц [41–43 и др.]. Рост устойчивости глицинатных и глицилглицинатных комплексов в водно-органических растворах (рис. 1, 2) значительно выше и соизмерим с изменением устойчивости комплексов с ацетат-ионом [44, 45 и др.].
Рис. 1.
Влияние состава водно-органических растворителей на устойчивость глицинатных комплексов Cu2+ и Ni2+ (а), Zn2+ и Mn2+ (б), Ag+ (в).
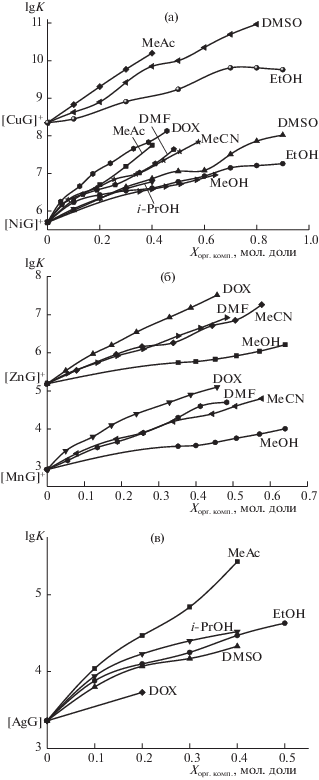
Рис. 2.
Влияние состава водно-органических растворителей на устойчивость глицилглицинатных комплексов Cu2+, Ni2+, Co2+.
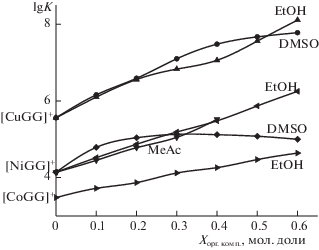
В водных растворах соотношение констант устойчивости комплексов двухзарядных ионов с глицинат-ионом, либо с глицилглицинат-ионом соответствует ряду Ирвинга–Вильямса: Mn2+ < < Co2+ < Ni2+ < Cu2+ > Zn2+. Природа органического сорастворителя оказывает дифференцирующее действие на устойчивость комплексных соединений, но соотношение констант устойчивости как глицинатов, так и глицилглицинатов d‑металлов в соответствующих водно-органических смесях при любой концентрации органического сорастворителя сохраняется по ряду Ирвинга–Вильямса (рис. 1, 2). Устойчивость глицината Ag+ занимает промежуточное положение между глицинатами марганца(II) и никеля(II) как в водных, так и в водно-органических растворах (рис. 1).
Изменение констант устойчивости комплексов d-металлов с лигандами аминного типа [43], ацетат- [44, 45], глицинат- (рис. 1) и глицилглицинат- (рис. 2) ионами в большинстве случаев описывается монотонно возрастающими зависимостями $\lg K$ = ƒ(Xорг.комп). В смесях воды с сильнокоординирующими растворителями (например, диметилсульфоксидом) для комплексов с аммиаком и этилендиамином зависимости $\lg K$ = = ƒ(Xорг.комп) носят экстремальный с максимумом характер [5, 41, 42]. Для глицинатов двухзарядных переходных металлов (рис. 1), так же как ацетатов [45], изменение $\lg K$ практически во всей области составов водно-диметилсульфоксидного растворителя прямолинейно. Для комплексов, в которых отсутствует координация по карбоксилатной группе, а именно глицилглицинатах никеля(II) и меди(II), глицинатах Ag(I), так же как для аминных комплексов, наибольший прирост констант устойчивости наблюдается в области низких концентраций диметилсульфоксида в растворе, при высоком содержании DMSO устойчивость глицилглицината меди(II) и глицината серебра(I) растет незначительно, а в случае глицилглицината никеля(II) даже несколько снижается (рис. 1, 2). В работе [6] было показано, что такое разнообразие характера зависимостей констант устойчивости координационных соединений от состава водно-органического растворителя определяется динамикой сольватационных вкладов реагентов в изменение энергии Гиббса реакций.
Используя значения констант устойчивости глицинатных и глицилглицинатных комплексов переходных металлов в водных (Kw) и водно-органических (${{K}_{{{\text{w}} + {\text{s}}}}}$) растворах [11–24], рассчитали изменение энергии Гиббса реакций при переходе от воды к ее смесям с органическим сорастворителем:
Рис. 3.
Влияние состава растворителей H2O–DMSO (а, б), H2O–EtOH (в, г), H2O–MeOH (д), H2O–iPrOH (е) на изменение энергии Гиббса пересольватации реагентов (Me, L) и реакций образования (r(MeL)) глицинатных (а, в, д, е) и глицилглицинатных (б, г) комплексов d-металлов (${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r([MeL])}}}}^{{^{ \circ }}}$).
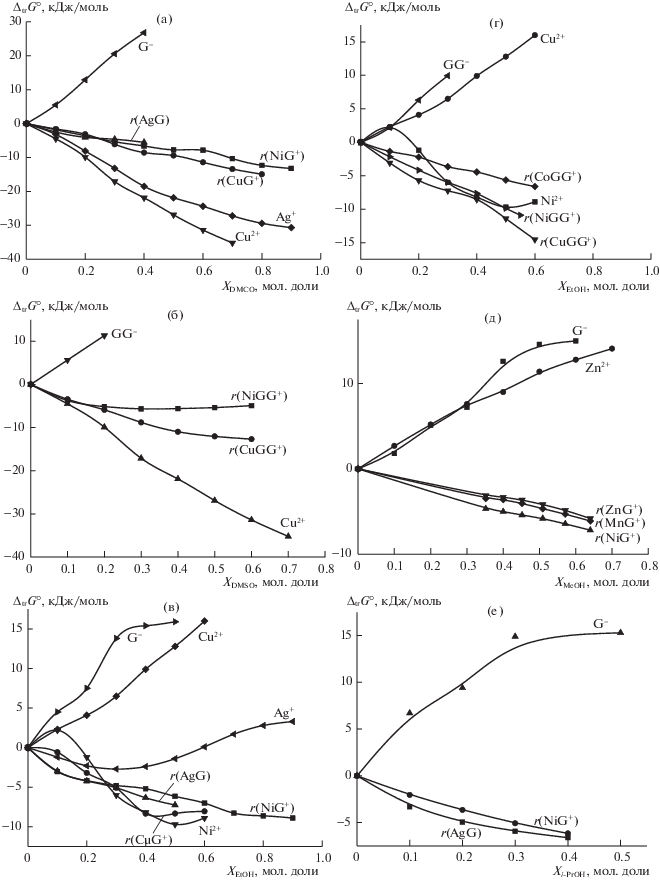
Установлено [5, 6], что монотонный рост устойчивости комплексов во всей области составов смешанного растворителя наблюдается в тех случаях, когда увеличение содержания органического сорастворителя приводит к ослаблению сольватации лиганда, а различие в изменении сольватации центрального и комплексного ионов невелико. Такая ситуация реализуется, например, при образовании комплексов d-металлов с лигандами аминного типа, ацетат-ионом [5, 6], а также с глицинат- и глицилглицинат-ионами, в водно-спиртовых растворах (рис. 3). В смесях с сильнокоординирующими растворителями, такими как диметилсульфоксид, изменение устойчивости комплексов с d-металлами зависит от действия двух сольватационных вкладов противоположной направленности. Ослабление сольватации лигандов способствует упрочнению их комплексов с ионами металлов, а стабилизация сольватной оболочки центрального иона в ДMSO дает противоположный вклад в смещение равновесия комплексообразования. Преобладание первого фактора в начальной области составов водно-диметилсульфоксидного растворителя, а при более высоком содержании DMSO – второго, обусловливает экстремальный (с максимумом) характер зависимости от состава растворителя вода–DMSO констант устойчивости комплексов d-металлов с аминами [5, 6]. Можно полагать, что аналогичные факторы обусловливают характер изменения устойчивости в водно-диметилсульфоксидных растворах глицилглицинатных комплексов d-металлов (рис. 3).
Термохимическое изучение реакций комплексообразования ионов d-металлов с глицинат- и глицилглицинат-ионами показало, что протекание данных процессов в водном растворе характеризуется высоким экзо-эффектом (∆Нr = –17.6 [28], –24.6 [29], –18.3 [26] и –16.7 кДж/моль [30] для комплексов [AgG], [CuG]+, [NiG]+ и [NiGG]+ соответственно (µ = 0.1 M)), сопоставимым с изменением энтальпии образования аммиачных комплексов (∆Нr = –16.2 [47] и –19.6 кДж/моль [48] для [NiNH3]2+ и [CuNH3]2+ соответственно (µ = 0.1M)).
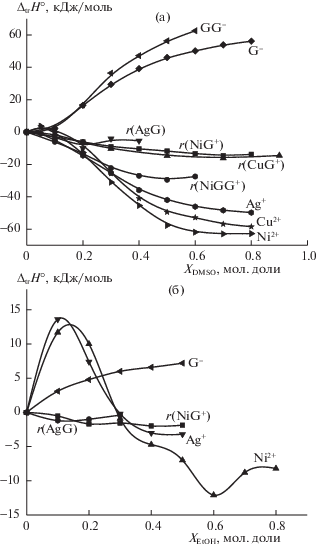
При переходе к водно-органическим растворителям наблюдается увеличение экзотермичности реакций образования глицинатных и глицилглицинатных комплексов d-металлов (рис. 4). Изменение ΔН° сольватации ионов металлов в большинстве случаев не способствует росту экзотермичности реакций при переходе от воды к смешанным растворителям. Изменение энтальпии пересольватации глицинат- и глицилглицинат-ионов положительно во всей области составов водно-органических растворителей, в значительной мере определяет рост экзо-эффекта реакций комплексообразования (рис. 4), но в большей или меньшей степени компенсируется различиями в сольватации комплексного и центрального ионов:
Рис. 4.
Влияние состава растворителей H2O–DMSO (а), H2O–EtOH (б) на изменение энтальпии пересольватации реагентов (Me, L) и реакций образования (r(MeL)) глицинатных и глицилглицинатных комплексов d-металлов.
В работе [6] показано, что термодинамические характеристики реакции комплексообразования (∆trY° = ∆trG°, ∆trH°) можно представить в виде функции двух переменных, а именно коэффициента различий (αdif) и изменения термодинамического состояния лиганда:
Таблица 1.
Значения коэффициентов различий для реакций образования комплексов d-металлов с глицинат-ионом и глицилглицинат-ионом в водно-органических растворах
| Реакция | Растворитель | ${{\alpha }_{{{\text{dif}}}}} = \frac{{{{\Delta }_{{{\text{tr}}}}}G_{{\text{r}}}^{^\circ }}}{{{{\Delta }_{{{\text{tr}}}}}G_{{\text{L}}}^{^\circ }}} + 1$ | ${{\alpha }_{{{\text{dif}}}}} = \frac{{{{\Delta }_{{{\text{tr}}}}}H_{{\text{r}}}^{^\circ }}}{{{{\Delta }_{{{\text{tr}}}}}H_{{\text{L}}}^{^\circ }}} + 1$ |
|---|---|---|---|
| Ni2+ + G− ⇄ [NiG]+ | H2O – EtOH | 0.7 (0.4) | 0.7 (0.5) |
| Ni2+ + G− ⇄ [NiG]+ | H2O – MeOH | 0.7 (0.4) | – |
| Ni2+ + G− ⇄ [NiG]+ | H2O – DMSO | 0.8 (0.4) | 0.8 (0.8) |
| Ni2+ + G− ⇄ [NiG]+ | H2O – iPrOH | 0.6 (0.4) | – |
| Cu2+ + G− ⇄ [CuG]+ | H2O – EtOH | 0.7 (0.4) | 0.7 (0.7) |
| Cu2+ + G− ⇄ [CuG]+ | H2O – DMSO | 0.7 (0.4) | – |
| Mn2+ + G− ⇄ [MnG]+ | H2O – MeOH | 0.7 (0.4) | – |
| Zn2+ + G− ⇄ [ZnG]+ | H2O – MeOH | 0.7 (0.4) | – |
| Ag+ + G− ⇄ [AgG] | H2O – EtOH | 0.6 (0.4) | 0.8 (0.2) |
| Ag+ + G− ⇄ [AgG] | H2O – iPrOH | 0.6 (0.4) | – |
| Ag+ + G− ⇄ [AgG] | H2O – DMSO | 0.8 (0.4) | 0.8 (0.4) |
| Ni2+ + GG− ⇄ [NiGG]+ | H2O – EtOH | 0.4 (0.3) | – |
| Ni2+ + GG− ⇄ [NiGG]+ | H2O – DMSO | 0.5 (0.2) | 0.5 (0.5) |
| Cu2+ + GG− ⇄ [CuGG]+ | H2O – EtOH | 0.3 (0.3) | – |
| Cu2+ + GG− ⇄ [CuGG]+ | H2O – DMSO | 0.5 (0.2) | – |
| Co2+ + GG− ⇄ [CoGG]+ | H2O – EtOH | 0.6 (0.3) | – |
Было рассчитано значение αdif для процесса образования комплекса меди(II) с диглицилглицинат ионом (GGG–) в водно-этанольном и водно-диметилсульфоксидном растворах. Значения ∆G этой реакции взяты из работ [49, 50]. Изменение энергии Гиббса пересольватации диглицилглицинат-иона в водно-этанольных смесях рассчитали, используя данные по растворимости диглицилглицина [51], значения ∆trG° протона [31], ∆Gr диссоциации диглицилглицина [52] в данном растворителе. Изменение энергии Гиббса пересольватации диглицилглицинат-иона в водно-диметилсульфоксидных смесях рассчитали, исходя из значений ∆trG° диглицилглицина [53], ∆trG° протона [31], ∆Gr диссоциации диглицилглицина [50] в данном растворителе.
Результаты расчетов показали, что значение αdif для реакции образования комплекса меди(II) с диглицилглицинат-ионом в водных растворах этанола и диметилсульфоксида составляет 0.5 и 0.6 соответственно. Полагаем, что для глицилглицинатных, а также диглицилглицинатных комплексов d-металлов является общим диапазон значений αdif = 0.3–0.6. Можно отметить, что в отличие от аминных комплексов [5, 6], для глицинатных и глицилглицинатных комплексов наблюдается совпадение (табл. 1) значений αdif, рассчитанных через изменение энергии Гиббса и энтальпийные характеристики реакций и сольватации лигандов.
В работе [6] на основе сопоставления значений αdif для процессов образования аминных, ацетатных и некоторых глицинатных комплексов сделан вывод о том, что увеличение коэффициента различий, а, следовательно, и увеличение различия в изменении сольватации центрального и комплексного ионов, происходит при переходе от нейтральных лигандов к заряженным и при увеличении размеров лиганда. Результаты данной работы показывают, что для заряженных N–O-донорных лигандов, образующих комплексы посредством атомов амино- и пептидной групп, диапазон значений коэффициентов различий соответствует установленным для комплексов с нейтральными лигандами аминного типа. Не происходит и увеличения значений αdif при образовании комплексов в ряду [MeG]−–[MeGG]−–[MeGGG]−. При переходе от глицинатных к глицилглицинатным комплексам удаленность карбоксилатной группы и ее неучастие в координации приводят к уменьшению коэффициента различий до значений, соответствующих аминным комплексам. В пептидных комплексах с увеличением длины лиганда величина αdif практически не изменяется, вероятно, в виду того, что в глицилглицинатных и диглицилглицинатных комплексах тип координации одинаков.
Таким образом, на примере образования глицинатных и глицилглицинатных комплексов d‑металлов, показана применимость общих закономерностей в изменении термодинамических параметров реакций в водно-органических растворах, установленных для аминных и ацетатных комплексов. Выявлены различия в долевом вкладе пересольватации глицинат- и глицилглицинат-иона в изменение термодинамических параметров реакций в смешанных растворителях, что позволило расширить возможности сольватационного подхода к описанию роли растворителя в реакциях комплексообразования с лигандами, имеющими пептидную группу.
Список литературы
Комов В.П., Шведова В.Н. Биохимия: учеб. для вузов. М.: Дрофа, 2004. 638 с.
Кузнецова Е.Г., Рыжикова В.А., Саломатина Л.А., Севастьянов В.И. // Вестник трансплантологии и искусственных органов. 2016. Т. 18. № 2. С. 152. https://doi.org/10.15825/1995-1191-2016-2-152-162
Гуля А.П., Граур В.О., Чумаков Ю.М. и др. // Журн. общ. химии. 2020. Т. 90. № 1. С. 143. https://doi.org/10.31857/S0044460X20010187
Ткачева А.Р., Шарутин В.В., Шарутина О.К., Слепухин П.А. // Там же. 2019. Т. 89. № 9. С. 1414. Tkacheva A.R., Sharutin V.V., Sharutina O.K., Slepukhin P.A. // Russ. J. Gen. Chem. 2019. V. 89. № 9. P. 1816. https://doi.org/10.1134/S1070363219090147
Шарнин В.А. // Там же. 1999. Т. 69. № 9. С. 1421. Sharnin V.A. // Russ. J. Gen. Chem. 1999. V. 69. № 9. P. 1368.
Шарнин В.А. // Изв. вузов. Химия и хим. технология. 2005. Т. 48. № 7. С. 44.
Shimazaki Y., Takani M., Yamauchi O. // Dalton Trans. 2009. V. 38. P. 7854. https://doi.org/10.1039/B905871K
Kiss T., Sovago I., Gergely A. // Pure and Appl. Chem. 1993. V. 65. № 5. P. 1029. https://doi.org/10.1351/pac199365051029
Эйхгорн Г. Неорганическая химия. Т. 1. М.: Мир, 1978. 713 с.
Datta S.P., Rabin B.R. // Trans. Faraday Soc. 1956. V. 52. P. 1117. https://doi.org/10.1039/TF9565201117
Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Журн. неорган химии. 1997. Т. 42. № 7. С. 1224.
Исаева В.А., Леденков С.Ф., Шарнин В.А., Шорманов В.А. // Коорд. химия. 1995. Т. 21. № 5. С. 396.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Там же. 1999. Т. 25. № 12. С. 912; Isaeva V.A., Sharnin V.A., Shormanov V.A. // Russ. J. Coord. Chem. 1999. V. 25. № 12. P. 852.
Исаева В.А., Шарнин В.А., Ганичева Н.В. // Журн. физ. химии. 2003. Т. 77. № 7. С. 1330. Isaeva V.A., Sharnin V.A., Ganicheva N.V. // Russ. J. Phys. Chem. A. 2003. V. 77. № 7. P. 1194.
Исаева В.А., Наумов В.В., Гессе Ж.Ф., Шарнин В.А. // Коорд. химия. 2008. Т. 34. № 8. С. 631. Isaeva V.A., Naumov V.V., Gesse Z.F., Sharnin V.A. // Russ. J. Coord. Chem. 2008. V. 34. № 8. P. 624. https://doi.org/10.1134/S1070328408080113
Исаева В.А., Гессе Ж.Ф., Наумов В.В., Шарнин В.А. // Журн. неорган химии. 2007. Т. 52. № 7. С. 1243. Isaeva V.A., Naumov V.V., Gesse Z.F., Sharnin V.A. // Russ. J. Inorg. Chem. 2007. V. 52. № 7. P. 1165. https://doi.org/10.1134/S0036023607070315
Mui K.-K., McBryde W.A.E., Nieboer E. // Canad. J. Chem. 1974. V. 52. № 10. P. 1821. https://doi.org/10.1139/v74-261
Ishiguro S., Suzuki H., Zama M. et al. // Bull. Chem. Soc. Jpn. 1986. V. 59. № 8. P. 2599. https://doi.org/10.1246/bcsj.59.2599
Исаева В.А., Наумов В.В., Шарнин В.А. // Коорд. химия. 2009. Т. 35. № 11. С. 878; Isaeva V.A., Naumov V.V., Sharnin V.A. // Russ. J. Coord. Chem. 2009. V. 35. P. 868. https://doi.org/10.1134/S107032840911013X
Наумов В.В., Исаева В.А., Шарнин В.А. // Журн. неорган химии. 2011. Т. 56. № 7. С. 1208. Naumov V.V., Isaeva V.A., Sharnin V.A. // Russ. J. Inorg. Chem. 2011. V. 56. № 7. P. 1139. https://doi.org/10.1134/S0036023611070199
Наумов В.В., Исаева В.А., Ковалева Ю.А., Шарнин В.А. // Журн. физ. химии. 2013. Т. 87. № 7. С. 1160. Naumov V.V., Isaeva V.A., Kovaleva Y.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2013. V. 87. № 7. P. 1135. https://doi.org/10.1134/S0036024413070224
Исаева В.А., Молчанов А.С., Кипятков К.А., Шарнин В.А. // Там же. 2019. Т. 93. № 8. С. 1164. Isaeva V.A., Molchanov A.S., Kipyatkov K.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2019. V. 93. № 8. P. 1460. https://doi.org/10.1134/S0036024419080107
Исаева В.А., Молчанов А.С., Кипятков К.А., Шарнин В.А. // Там же.2020. Т. 94. № 2. С. 182. Isaeva V.A., Molchanov A.S., Kipyatkov K.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2020. V. 94. № 2. P. 182. https://doi.org/10.31857/S0044453720020132
Исаева В.А., Молчанов А.С., Шишкин М.В. и др. // Журн. неорган. химии. 2020. Т. 65. № 4. С. 517. Isaeva V.A., Molchanov A.S., Shishkin M.V., Kipyatkov K.A., Sharnin V.A. // Russ. J. Inorg. Chem. 2020. V. 65. № 4. P. 535. https://doi.org/10.1134/S0036023620040075
Леденков С.Ф., Шорманов В.А., Шарнин В.А. // Журн. физ. химии. 1996. Т. 70. № 10. С. 1769. Ledenkov S.F., Shormanov V.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 1996. V. 70. № 10. P. 1642.
Михеев С.В., Шарнин В.А. // Там же. 2010. Т. 84. № 2. С. 205. Mikheev S.V., Sharnin V.A. // Russ. J. Phys. Chem. A. 2010. V. 84. № 2. P. 153. https://doi.org/10.1134/S0036024410020019
Gesse Zh.F., Repkin G.I., Isaeva V.A., Sharnin V.A. // J. Therm. Anal. Calorim. 2012. V. 110. № 3. P. 1457. https://doi.org/10.1007/s10973-011-2127-z
Гессе Ж.Ф., Исаева В.А., Репкин Г.И., Шарнин В.А. // Журн. физ. химии. 2012. Т. 86. № 1. С. 59. Gesse Zh.F., Isaeva V.A., Repkin G.I., Sharnin V.A. // Russ. J. Phys. Chem. A. 2012. V. 86. № 1. P. 53. https://doi.org/10.1134/S0036024412010104
Михеев С.В., Спичко А.В., Шарнин В.А. // Там же. 2012. Т. 86. № 2. С. 272. Mikheev S.V., Spichko A.V., Sharnin V.A. // Russ. J. Phys. Chem. A. 2012. V. 86. № 2. P. 215. https://doi.org/10.1134/S0036024412010220
Наумов В.В., Ковалева Ю.А., Исаева В.А. и др. // Там же. 2014. Т. 88. № 6. С. 969. Naumov V.V., Kovaleva Y.A., Isaeva V.A., Usacheva T.R., Sharnin V.A. // Russ. J. Phys. Chem. A. 2014. V. 88. № 6. P. 932. https://doi.org/10.1134/S0036024414060193
Kalidas C., Hefter G., Marcus Y. // Chem. Rev. 2000. V. 100. № 3. P. 819. https://doi.org/10.1021/cr980144k
Hefter G., Marcus Y., Waghorne W.E. // Ibid. 2002. V. 102. № 8. P. 2773. https://doi.org/10.1021/cr010031s
Невский А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Коорд. химия. 1983. Т. 9. № 3. С. 391.
Шорманов В.А., Шарнин В.А., Леденков С.Ф. // Журн. физ. химии. 1996. Т. 70. № 8. С. 1521; Shormanov V.A., Sharnin V.A., Ledenkov S.F. // Russ. J. Phys. Chem. A. 1996. V. 70. № 8. P. 1418.
Исаева В.А., Наумов В.В., Шарнин В.А. // Там же. 2016. Т. 90. № 5. С. 721. Isaeva V.A., Naumov V.V., Sharnin V.A. // Russ. J. Phys. Chem. A. 2016. V. 90. № 5. P. 973. https://doi.org/10.1134/S0036024416050162
Наумов В.В., Исаева В.А., Шарнин В.А. // Там же. 2014. Т. 88. № 3. С. 443; Naumov V.V., Isaeva V.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2014. V. 88. № 3. P. 433. https://doi.org/10.1134/S0036024414030194
Dey B.P., Lahiri S.C. // Indian J. Chem. 1986. V. 25A. № 2. P. 136.
Исаева В.А., Молчанов А.С., Кипятков К.А., Шарнин В.А. // Журн. физ. химии. 2020. Т. 94. № 1. С. 16. Isaeva V.A., Molchanov A.S., Kipyatkov K.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2020. V. 94. № 1. P. 16. https://doi.org/10.31857/S0044453720010100
Гессе Ж.Ф., Исаева В.А., Шарнин В.А. // Там же. 2010. Т. 84. № 2. С. 385. Gesse Zh.F., Isaeva V.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2010. V. 84. № 2. P. 329. https://doi.org/10.1134/S0036024410020299
Наумов В.В., Исаева В.А., Кузина Е.Н., Шарнин В.А. // Там же. 2012. Т. 86. № 12. С. 1907. Naumov V.V., Isaeva V.A., Kuzina E.N., Sharnin V.A. // Russ. J. Phys. Chem. A. 2012. V. 86. № 12. P. 1773. https://doi.org/10.1134/S0036024412120175
Нищенков А.В., Шарнин В.А., Шорманов В.А., Крестов Г.А. // Коорд. химия. 1990. Т. 16. № 9. С. 1264.
Леденков С.Ф., Шарнин В.А., Исаева В.А., Шорманов В.А. // Там же. 1993. Т. 19. № 9. С. 691.
Невский А.В., Шорманов В.А., Крестов Г.А. // Там же. 1985. Т. 11. № 5. С. 666.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Журн. физ. химии. 1998. Т. 72. № 12. С. 2182. Isaeva V.A., Sharnin V.A., Shormanov V.A. // Russ. J. Phys. Chem. A. 1998. V. 72. № 12. P. 1985.
Исаева В.А., Шарнин В.А., Шорманов В.А., Щербина И.В. // Коорд. химия. 1998. Т. 24. № 2. С. 149; Isaeva V.A., Sharnin V.A., Shormanov V.A., Shcherbina I.V. // Russ. J. Coord. Chem. 1998. V. 24. № 2. P. 139.
Wells C.F. // Advances in Chemistry. 1979. V. 177. P. 53. https://doi.org/10.1021/ba-1979-0177.ch005
Шарнин В.А., Шорманов В.А., Марков В.Н., Крестов Г.А. // Коорд. химия. 1985. Т. 11. № 6. С. 778.
Михеев С.В., Шарнин В.А. // Журн. физ. химии. 2004. Т. 78. № 9. С. 1709. Mikheev S.V., Sharnin V.A. // Russ. J. Phys. Chem. A. 2004. V. 78. № 9. P. 1502.
Фам Тхи Л., Усачева Т.Р., Хренова Т.М., Шарнин В.А. // Там же. 2017. Т. 91. № 7. С. 1161; Pham Thi L., Usacheva T.R., Khrenova T.M., Sharnin V.A. // Russ. J. Phys. Chem. A. 2017. V. 91. № 7. P. 1235. https://doi.org/10.1134/S003602441707010X
Usacheva T.R., Pham Thi L., Kuzmina K.I., Sharnin V.A. // J. Therm. Anal. Calorim. 2017. V. 130. № 1. P. 471. https://doi.org/10.1007/s10973-017-6207-6
Jie Lu, Xiu-Juan Wang, Xia Yang, Chi-Bun Ching // J. Chem. Eng. Data. 2006. V. 51. № 5. P. 1593. https://doi.org/10.1021/je0600754
Фам Тхи Л., Усачева Т.Р., Тукумова Н.В. и др. // Журн. физ. химии. 2016. Т. 90. № 2. С. 216; Pham Tkhi L., Usacheva T.R., Tukumova N.V., Koryshev N.E., Khrenova T.M., Sharnin V.A. // Russ. J. Phys. Chem. A. 2016. V. 90. № 2. P. 344. https://doi.org/10.1134/S0036024416020138
Усачева Т.Р., Кузьмина К.И., Лан Фам Тхи и др. // Там же. 2014. Т. 88. № 7−8. С. 1176; Usacheva T.R., Kuzmina K.I., Lan Pham Tkhi, Kuzmina I.A., Sharnin V.A. // Russ. J. Phys. Chem. A. 2014. V. 88. № 8. P. 1357. https://doi.org/10.1134/S0036024414080305
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии