

Журнал физической химии, 2021, T. 95, № 7, стр. 1018-1026
Структурно-термодинамические характеристики малеиновой кислоты в водно-органических растворителях
Н. В. Тукумова a, *, Н. В. Белова a, Т. Р. Усачева a, Чан Тхи Зьеу Тхуан b, **
a Ивановский государственный химико-технологический университет
153000 Иваново, Россия
b Индустриальный университет города ХоШиМина
Го Вап, Вьетнам
* E-mail: oxt@isuct.ru
** E-mail: tranthidieuthuan@iuh.edu.vn
Поступила в редакцию 02.08.2020
После доработки 02.08.2020
Принята к публикации 10.11.2020
Аннотация
Представлены результаты изучения структуры малеиновой кислоты, процессов газофазного протонирования, а также кислотно-основных равновесий с участием малеиновой кислоты в водно-этанольных и водно-диметилсульфоксидных растворах. Определены величины рK диссоциации малеиновой кислоты в водно-этанольных и водно-диметилсульфоксидных растворителях. Установлено, что с ростом содержания неводного компонента в водно-органической смеси значения рK увеличиваются. Анализ сольватационных вкладов всех реагентов, участвующих в процессе кислотной диссоциации малеиновой кислоты в водно-этанольных и водно-диметилсульфоксидных растворителях, показал, что основной вклад в увеличение значений рK1 и рK2 вносит десольватация анионов.
Малеиновая кислота относится к широко распространенным биолигандам и входит в состав многих лекарственных препаратов. Анализ литературных источников показывает, что интерес к исследованиям процессов с участием малеиновой кислоты и практическим аспектам ее применения в различных наукоемких технологиях выходит за рамки традиционного применения в фармацевтике. Например, малеиновая кислота используется в качестве эффективного лиганда для связывания цинка, никеля, циркония в координированных полимерах при создании новых наноматериалов для катализа [1] и при активации регенерированных CoMo/Al2O3 катализаторов [2]. Исследования термодинамического поведения малеиновой кислоты необходимы для разработки селективных хемосенсоров для ее определения в смеси органических компонентов и дальнейшего выделения из растворов [3] и при исследовании модельных процессов транспорта через липидные мембраны наночастиц полимеров, содержащих малеиновую кислоту [4]. Для эффективного использования малеиновой кислоты в составе наноматериалов необходима информация о структуре и термодинамических свойствах малеиновой кислоты в различных средах.
Повышенное внимание исследователей привлекают процессы депротонирования, в том числе газофазного. Энергия газофазного депротонирования является отражением кислотности соединения в отсутствие влияния среды. Процессы диссоциации кислоты в газовой фазе H2L ↔ HL– + + H+ (HL– ↔ L2– + H+) сопровождаются изменением энтальпии и энергии Гиббса [5–12]. Анализ изменения этих величин позволяет выделить эффекты, непосредственно связанные только с влиянием конформационных особенностей или введения заместителей на кислотность соединений, не искаженные действием среды (см., например, [13]). Определение энергий газофазного депротонирования позволяет выстроить шкалу кислотности, выявить соединения со сверхкислыми свойствами, установить связь энергий депротонирования с величинами pKa. Авторы [14] ввели классификацию кислот, согласно которой кислота считается сверхсильной, если энергия Гиббса газофазного депротонирования менее 300 ккал/моль.
Настоящая работа продолжает исследования, начатые в работах [15–17], и представляет результаты изучения структуры малеиновой кислоты, процессов газофазного протонирования, а также кислотно-основных равновесий с участием малеиновой кислоты в водно-этанольных и водно-диметилсульфоксидных растворах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для нахождения величин рK диссоциации малеиновой кислоты в водно-органических растворах использовали метод потенциометрического титрования, основанный на измерении разности потенциалов между стеклянным индикаторным электродом и хлорсеребряным электродом сравнения. Исследования проводили при ионной силе 0.1 на фоне перхлората натрия при температуре 298 К. В качестве титранта использовали безкарбонатный раствор гидроксида натрия с концентрацией 0.1 М, аналогичный по содержанию органического компонента раствору в ячейке. Содержание этанола в растворах изменялось от 0 до 0.7 мол. доли, диметилсульфоксида от 0 до 0.3 мол. доли. Ограничение содержания органических растворителей в водно-органических растворах вызвано ухудшением растворимости в нем, как малеиновой кислоты, так и щелочи. Подробное описание методики проведения потенциометрического титрования приведено в работах [15–17].
Математическую обработку полученных в ходе потенциометрического титрования данных, проводили по универсальной программе “PHMETR”, предназначенной для расчета равновесий с произвольным числом реакций в растворе, алгоритм которой приведен в [18].
Стехиометрическая модель системы учитывала следующие равновесия:
(1)
${{{\text{Н}}}_{{\text{2}}}}{\text{L}} \leftrightarrow {{{\text{H}}}^{ + }} + {\text{Н}}{{{\text{L}}}^{ - }},$(4)
${{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {{{\text{H}}}^{ + }} + {\text{O}}{{{\text{H}}}^{ - }}.$Значение константы равновесия процесса (4), приведенное в работе [21] при нулевой ионной силе, было также пересчитано на ионную силу 0.1 по уравнению с одним индивидуальным параметром [20] с учетом процесса ионизации воды в водно-органических растворителях [22, 23].
Анализ сольватационных вкладов участников реакций кислотной диссоциации малеиновой кислоты при переносе из воды в водно-органические смеси проводили c использованием уравнений:
(5)
$\begin{gathered} {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1}}}}^{0} = {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} - {\text{орг}}{\text{.комп}})}}^{0}--{{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1(}}{{{\text{H}}}_{{\text{2}}}}{\text{O)}}}}^{0} = \\ = [{{\Delta }_{{{\text{tr}}}}}{{G}^{0}}({\text{H}}{{{\text{L}}}^{ - }})--{{\Delta }_{{{\text{tr}}}}}{{G}^{0}}({{{\text{H}}}_{{\text{2}}}}{\text{L}})] + {{\Delta }_{{{\text{tr}}}}}{{G}^{0}}({{{\text{H}}}^{ + }}), \\ \end{gathered} $(6)
$\begin{gathered} {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2}}}}^{0} = {{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} - {\text{орг}}{\text{.комп}})}}^{0}--{{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2(}}{{{\text{H}}}_{{\text{2}}}}{\text{O)}}}}^{0} = \\ = [{{\Delta }_{{{\text{tr}}}}}{{G}^{0}}({{{\text{L}}}^{{2 - }}})--{{\Delta }_{{{\text{tr}}}}}{{G}^{0}}({\text{H}}{{{\text{L}}}^{ - }})] + {{\Delta }_{{{\text{tr}}}}}{{G}^{0}}({{{\text{H}}}^{ + }}), \\ \end{gathered} $ΔtrG0(H+) – величины изменения энергии Гиббса переноса протона из воды в водно-этанольные [24] и водно-диметилсульфоксидные растворы [25]; ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1(}}{{{\text{H}}}_{{\text{2}}}}{\text{O)}}}}^{0}$, ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2(}}{{{\text{H}}}_{{\text{2}}}}{\text{O)}}}}^{0}$, ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} - {\text{орг}}{\text{.комп}})}}^{0}$, ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2(}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} - {\text{орг}}{\text{.комп}})}}^{0}$ – значения изменения энергии Гиббса для первой и второй ступеней диссоциации малеиновой кислоты в воде и в водно-органических смесях, соответственно.
Значения ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1}}}}^{0}$ и ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2}}}}^{0}$ для процессов (5) и (6) рассчитывали по уравнению:
где $\lg {{K}_{i}}$ – логарифмы констант диссоциации малеиновой кислоты по первой (1) и второй (2) ступени.Квантово-химические расчеты в данной работе выполнялись с использованием пакета программ GAUSSIAN 03 [26] в приближении теории функционала электронной плотности (функционал B3LYP [27–31]). Во всех расчетах использовались корреляционно-согласованные валентно-трехэкспонентные базисы cc-pVTZ [32]. Для каждой рассмотренной конфигурации проведена оптимизация геометрических параметров с последующим вычислением матрицы вторых производных полной энергии по декартовым координатам ядер. Изучение распределения электронной плотности в молекулах проводилось в рамках анализа натуральных орбиталей (NBO) с помощью программы NBO 3.1 [33], входящей в состав программного комплекса GAUSSIAN 03. Визуализация полученных структур выполнена с помощью программы ChemCraft [34]. Изучение строения изомеров и энергий депротонирования в сольватированном состоянии выполнено в рамках модели реактивного поля PCM [35].
При исследовании процесса депротонирования по первой ступени (уравнение (1)) энергии диссоциации изомеров ΔrЕ оценивали как разность электронных энергий депротонированных форм кислот Е(HL–) и энергий их молекулярных форм Е(H2L): ΔrЕ = Е(НL–) – Е(Н2L). При этом принимали: $E_{{{{{\text{H}}}^{ + }},0}}^{0}$ = 0.
Величины ${{\Delta }_{{\text{r}}}}H_{{298}}^{0}$, ${{\Delta }_{{\text{r}}}}G_{{298}}^{0}$ рассчитывали по уравнениям :
(8)
$\begin{gathered} {{\Delta }_{{\text{r}}}}H_{{298}}^{0} = H_{{298}}^{0}({\text{H}}{{{\text{L}}}^{ - }})--H_{{298}}^{0}({{{\text{H}}}_{2}}{\text{L}}) + \\ + \;1.48,\;{\text{ккал/моль}}, \\ \end{gathered} $(9)
$\begin{gathered} {{\Delta }_{{\text{r}}}}G_{{298}}^{0} = G_{{298}}^{0}({\text{H}}{{{\text{L}}}^{ - }})--G_{{298}}^{0}\left( {{{{\text{H}}}_{{\text{2}}}}{\text{L}}} \right)-- \\ - \;6.27,\;{\text{ккал/моль}}{\text{.}} \\ \end{gathered} $Расчеты для диссоциации по второй ступени проводили подобным образом.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
На рис. 1 представлены конфигурации малеиновой кислоты, соответствующие минимумам на поверхности потенциальной энергии, а также их относительные энергии в газовой фазе и в водном растворе. Наиболее низкой относительной энергией обладает транс-изомер (t1). Относительное содержание изомеров в газовой фазе, рассчитанное на основании теоретических значений относительных энергий Гиббса, ΔG0, составляет 76.0%, 23.0%, 0.2%, 0.8% для конфигураций t1, t2, c1 и c2 соответственно. Наличие двойной связи С2=С3 обеспечивает плоскостность углерод-кислородного каркаса, в результате все четыре изомера в газовой фазе обладают симметрией CS. В водном растворе незначительно увеличивается содержание цис-изомеров: 57.0%, 40.0%, 2.3%, 0.2% для конфигураций t1, t2, c1 и c2 соответственно. Причем, если для первых трех изомеров, как и в газовой фазе, минимуму на ППЭ соответствует конформация с плоским строением, для изомера с2 структура симметрии СS является седловой точкой первого порядка. Форма колебаний, отвечающая мнимой частоте, соответствует отклонению от плоскостности углерод-кислородного каркаса. Движение по этой координате позволило найти минимум на ППЭ.
Рис. 1.
Стабильные цис-, (c), и транс-, (t), конфигурации малеиновой кислоты, соответствующие минимумам на ППЭ, и их относительные энергии газовой фазе – ΔE и в водном растворе – ΔEw (ккал/моль) по данным квантово-химических расчетов (B3LYP/cc-pVTZ).
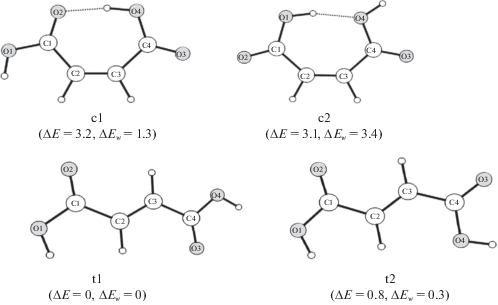
В табл. 1 представлены основные геометрические параметры равновесных структур. В табл. 2–4 сведены результаты анализа распределения электронной плотности. Таблица 2 представляет величины натуральных зарядов на атомах, табл. 3–индексы Вайберга, которые могут служить характеристикой порядков связей. В рамках схемы NBO перераспределение электронной плотности в результате донорно-акцепторного взаимодействия между заполненными натуральными орбиталями (Льюисового типа) и формально незанятыми орбиталями молекулы приводит к стабилизации. В табл. 5 представлены наибольшие рассчитанные значения энергий взаимодействия по типу “донор–акцептор” E(2), полученные в NBO-анализе с использованием теории возмущения второго порядка для изученных структур. Во всех изомерах малеиновой кислоты наблюдается гиперсопряжение между связывающей орбиталью π(С2–С3) и разрыхляющими орбиталями π*(С1–О2) и π*(С4–О3), что приводит к понижению энергии и дополнительной стабилизации молекулярной системы. Наличие гиперсопряжений в углерод-кислородном каркасе приводит к изменению соответствующих межъядерных расстояний (табл. 1), а также индексов Вайберга (табл. 3) по сравнению с величинами, характерными для одинарных или двойных связей. Кроме того, наличие взаимодействия неподеленной пары атома кислорода с σ*(O–H) в цис-изомерах является доказательством наличия водородной связи, о чем свидетельствуют и величины межъядерных расстояний r(O⋅⋅⋅O), которые меньше суммы ван-дер-ваальсовых радиусов атомов кислорода (rв–д–в(O) = 1.4 Å), а также – значения r(O⋅⋅⋅Н). Интересно отметить, что согласно результатам расчетов, в водном растворе во всех изомерах происходит усиление сопряжения в углерод-кислородной цепи, а также немного увеличивается сила водородной связи в цис-изомерах.
Таблица 1.
Основные геометрические параметры изомеров малеиновой кислоты по данным квантово-химических расчетов (B3LYP/cc-pVTZ)
| Связь | Изомер (t1) | Изомер (t2) | Изомер (c1) | Изомер (c2) | ||||
|---|---|---|---|---|---|---|---|---|
| газ | раствор | газ | раствор | газ | раствор | газ | раствор | |
| Межъядерные расстояния, Å | ||||||||
| C1–C2 | 1.492 | 1.488 | 1.493 | 1.490 | 1.482 | 1.481 | 1.505 | 1.498 |
| C2–C3 | 1.329 | 1.329 | 1.330 | 1.330 | 1.339 | 1.338 | 1.339 | 1.338 |
| С3–С4 | 1.483 | 1.484 | 1.479 | 1.480 | 1.508 | 1.500 | 1.478 | 1.480 |
| C1–O1 | 1.356 | 1.347 | 1.357 | 1.347 | 1.344 | 1.332 | 1.334 | 1.330 |
| C1–O2 | 1.199 | 1.207 | 1.199 | 1.207 | 1.212 | 1.220 | 1.205 | 1.212 |
| C4–O3 | 1.206 | 1.209 | 1.205 | 1.210 | 1.207 | 1.214 | 1.202 | 1.205 |
| C4–O4 | 1.350 | 1.344 | 1.355 | 1.344 | 1.324 | 1.324 | 1.365 | 1.354 |
| C2–H | 1.084 | 1.083 | 1.083 | 1.082 | 1.085 | 1.083 | 1.083 | 1.082 |
| С3–Н | 1.081 | 1.081 | 1.081 | 1.081 | 1.082 | 1.082 | 1.082 | 1.082 |
| O1–H | 0.964 | 0.966 | 0.964 | 0.966 | 0.964 | 0.967 | 0.973 | 0.979 |
| О4–H | 0.969 | 0.970 | 0.968 | 0.969 | 0.989 | 0.999 | 0.969 | 0.970 |
| Н⋅⋅⋅О | 1.631 | 1.578 | 1.713 | 1.680 | ||||
| O⋅⋅⋅O | 2.610 | 2.569 | 2.668 | 2.639 | ||||
| Валентные углы, град. | ||||||||
| О1–С1–С2 | 115.3 | 115.8 | 115.3 | 116.0 | 115.6 | 116.3 | 121.0 | 121.4 |
| С1–С2–С3 | 121.1 | 121.1 | 120.7 | 120.6 | 128.4 | 128.5 | 136.1 | 135.0 |
| C2–C3–С4 | 120.7 | 121.0 | 124.4 | 124.4 | 133.8 | 133.0 | 133.6 | 133.1 |
| С3–С4–О4 | 111.1 | 111.1 | 113.5 | 113.8 | 120.2 | 120.3 | 115.9 | 115.6 |
| О1–C1–O2 | 120.5 | 119.8 | 120.4 | 119.8 | 118.5 | 118.1 | 121.1 | 120.2 |
| O3–C4–O4 | 123.3 | 123.5 | 122.9 | 123.3 | 122.4 | 121.2 | 121.2 | 122.0 |
| Н–О1–С1 | 111.0 | 111.8 | 111.1 | 111.9 | 111.5 | 112.3 | 111.8 | 112.0 |
| Н–С2–С1 | 119.9 | 119.0 | 119.3 | 118.8 | 115.3 | 115.2 | 108.3 | 109.2 |
| Н–С3–С4 | 118.6 | 117.9 | 115.3 | 115.0 | 109.5 | 110.4 | 109.6 | 110.3 |
| Н–О4–С4 | 107.2 | 108.3 | 106.7 | 108.1 | 111.9 | 111.4 | 107.8 | 109.6 |
| Торсионные углы, град. | ||||||||
| О1–С1–С2–С3 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 0.0 | –6.8 |
| C1–C2–C3–С4 | 180.0 | 180.0 | 180.0 | 180.0 | 0.0 | 0.0 | 0.0 | 0.4 |
| С2–С3–С4–О4 | 180.0 | 180.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 13.1 |
| О2–С1–С2–С3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 180.0 | 173.2 |
| С2–С3–С4–О3 | 0.0 | 0.0 | 180.0 | 180.0 | 180.0 | 180.0 | 180.0 | 167.7 |
| Н–О1–С1–С2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 |
| Н–О4–С4–С3 | 180.0 | 180.0 | 180.0 | 180.0 | 0.0 | 0.0 | 180.0 | 178.6 |
Таблица 2.
Натуральные заряды на атомах (в долях элементарного заряда) в изомерах малеиновой кислоты, полученные в результате квантово-химических расчетов (B3LYP/cc-pVTZ)
| Атомы | Изомер (t1) | Изомер (t2) | Изомер (c1) | Изомер (c2) | ||||
|---|---|---|---|---|---|---|---|---|
| газ | раствор | газ | раствор | газ | раствор | газ | раствор | |
| C1 | 0.750 | 0.765 | 0.750 | 0.765 | 0.769 | 0.783 | 0.734 | 0.750 |
| С2 | –0.253 | –0.246 | –0.263 | –0.253 | –0.316 | –0.281 | –0.205 | –0.219 |
| С3 | –0.226 | –0.221 | –0.217 | –0.216 | –0.175 | –0.192 | –0.284 | –0.256 |
| С4 | 0.749 | 0.761 | 0.745 | 0.758 | 0.732 | 0.748 | 0.749 | 0.762 |
| О1 | –0.654 | –0.661 | –0.654 | –0.661 | –0.634 | –0.634 | –0.660 | –0.669 |
| О2 | –0.541 | –0.596 | –0.539 | –0.596 | –0.577 | –0.613 | –0.537 | –0.597 |
| О3 | –0.578 | –0.607 | –0.551 | –0.594 | –0.543 | –0.603 | –0.540 | –0.571 |
| О4 | –0.657 | –0.658 | –0.680 | –0.670 | –0.655 | –0.672 | –0.712 | –0.700 |
| Н(О1) | 0.475 | 0.501 | 0.473 | 0.501 | 0.481 | 0.509 | 0.496 | 0.507 |
| Н(С2) | 0.212 | 0.225 | 0.208 | 0.226 | 0.200 | 0.227 | 0.225 | 0.230 |
| Н(С3) | 0.236 | 0.233 | 0.239 | 0.233 | 0.223 | 0.231 | 0.228 | 0.235 |
| Н(О4) | 0.487 | 0.503 | 0.490 | 0.506 | 0.496 | 0.498 | 0.507 | 0.526 |
Таблица 3.
Индексы Вайберга для связей в конформерах малеиновой кислоты по данным квантово-химических расчетов (B3LYP/cc-pVTZ)
| Изомер (t1) | Изомер (t2) | Изомер (c1) | Изомер (c2) | |||||
|---|---|---|---|---|---|---|---|---|
| газ | раствор | газ | раствор | газ | раствор | газ | раствор | |
| C1–О1 | 1.030 | 1.059 | 1.029 | 1.059 | 1.070 | 1.107 | 1.084 | 1.101 |
| С1–О2 | 1.769 | 1.719 | 1.772 | 1.719 | 1.672 | 1.620 | 1.740 | 1.694 |
| С1–С2 | 1.008 | 1.015 | 1.006 | 1.013 | 1.036 | 1.038 | 1.000 | 1.012 |
| С2–С3 | 1.860 | 1.857 | 1.856 | 1.854 | 1.852 | 1.852 | 1.857 | 1.859 |
| С3–С4 | 1.012 | 1.013 | 1.023 | 1.024 | 0.992 | 1.007 | 1.036 | 1.030 |
| С4–О3 | 1.741 | 1.714 | 1.755 | 1.714 | 1.722 | 1.681 | 1.774 | 1.746 |
| С4–О4 | 1.053 | 1.072 | 1.034 | 1.066 | 1.117 | 1.125 | 0.986 | 1.015 |
Таблица 4.
Энергии донорно-акцепторного взаимодействия в конформерах малеиновой кислоты (Е2, ккал/моль) по данным квантово-химических расчетов (B3LYP/cc-pVTZ)
| Связь | Изомер (t1) | Изомер (t2) | Изомер (c1) | Изомер (c2) | ||||
|---|---|---|---|---|---|---|---|---|
| газ | раствор | газ | раствор | газ | раствор | газ | раствор | |
| π(С2–С3) – π*(О2–С1) | 16.6 | 17.9 | 16.6 | 17.8 | 20.0 | 20.2 | 12.6 | 14.6 |
| π(С2–С3) – π*(О3–С4) | 17.5 | 17.8 | 17.5 | 18.1 | 11.3 | 14.0 | 18.7 | 17.6 |
| lp(O1) – π*(О2–С1) | 44.5 | 47.9 | 44.3 | 47.9 | 48.6 | 53.3 | 51.2 | 53.3 |
| lp(O4) – π*(O3–C4) | 47.1 | 49.5 | 45.2 | 49.2 | 56.1 | 57.0 | 39.8 | 42.7 |
| lp(O2) – σ*(O1–C1) | 34.6 | 32.0 | 34.9 | 32.0 | 31.4 | 28.1 | 31.8 | 29.9 |
| lp(O2) – σ*(C1–C2) | 21.7 | 20.4 | 21.6 | 20.5 | 14.2 | 12.0 | 21.0 | 19.3 |
| lp(O3) – σ*(O4–C4) | 32.8 | 31.5 | 33.4 | 31.4 | 30.0 | 28.7 | 35.4 | 33.5 |
| lp(O3) – σ*(C3–C4) | 21.1 | 20.5 | 20.1 | 19.2 | 21.7 | 19.8 | 19.8 | 19.3 |
| lp(O2) – σ*(O4–H) | 23.7 | 31.4 | ||||||
| lp(O4) – σ*(O1–H) | 15.5 | 17.7 | ||||||
Таблица 5.
Величины констант диссоциации малеиновой кислоты в смесях вода–этанол и вода–диметилсульфоксид
| Xр-ля, мол. доли | Вода–этанол | Вода–ДМСО | ||
|---|---|---|---|---|
| pK1 | pK2 | pK1 | pK2 | |
| 0 | 1.76 ± 0.04 | 5.78 ± 0.04 | 1.76 ± 0.04 | 5.78 ± 0.04 |
| 0.1 | 1.80 ± 0.04 | 5.90 ± 0.06 | 2.15 ± 0.08 | 7.31 ± 0.08 |
| 0.3 | 1.83 ± 0.08 | 6.53 ± 0.07 | 2.48 ± 0.12 | 8.90 ± 0.08 |
| 0.4 | 1.96 ± 0.04 | 7.50 ± 0.06 | – | – |
| 0.5 | 2.20 ± 0.07 | 7.89 ± 0.07 | – | – |
| 0.7 | 2.44 ± 0.08 | 8.38 ± 0.10 | – | – |
В таблице 5 представлены величины рK диссоциации в водно-этанольных и водно-диметилсульфоксидных растворах.
Следует отметить, что темпы роста констант диссоциации малеиновой кислоты в системе вода–ДМСО выше, чем в системе вода–этанол, что можно объяснить различием в величинах диэлектрической проницаемости этанола (24.3) и диметилсульфоксида (46.68). Отметим, что значения рK1 и рK2 диссоциации малеиновой кислоты сильно различаются друг от друга.
Значения ∆trG0(L2–) и ∆trG0(HL–) в литературе на данный момент отсутствуют. Зависимости величин изменения энергии Гиббса реагентов и реакции диссоциации малеиновой кислоты по первой и второй ступени от состава водно-этанольного и водно-диметилсульфоксидного растворителей представлены на рис. 2–5. Как видно из рис. 2–5 при переходе от воды к водно-органическим смесям рост величин ${{\Delta }_{{{\text{tr}}}}}G_{{\text{r}}}^{0}$ реакций кислотной диссоциации малеиновой кислоты определяется различиями в изменении сольватных состояний протонированной и депротонированной форм малеиновой кислоты по обеим ступеням диссоциации: [∆trG°(HL–) – ∆trG°(H2L)] и [∆trG°(L2–) – ‒ ∆trG°(HL–)]. Можно предположить, что увеличение значений рК диссоциации малеиновой кислоты с ростом содержания неводного компонента в растворах связано главным образом с дестабилизацией анионов.
Рис. 2
. Зависимости изменения энергии Гиббса реагентов реакции диссоциации малеиновой кислоты по первой ступени при переносе из воды в водные растворы этанола от состава водно-этанольного растворителя: 1 – [∆trG0(HL–) – ∆trG0(H2L)], 2 – ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r1}}}}^{0}$, 3 – ∆trG0(H+).
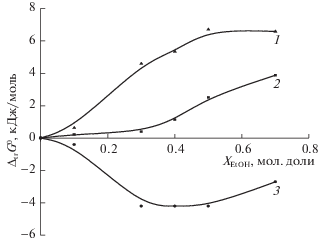
Рис. 3.
Зависимости изменения энергии Гиббса реагентов реакции диссоциации малеиновой кислоты по второй ступени при переносе из воды в водный раствор этанола от состава водно-этанольного растворителя: 1 – [∆trG0(L2–) – ∆trG0(HL–)], 2 – ∆trGr2, 3 – ∆trG(H+).
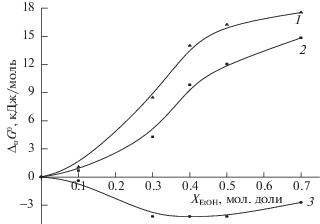
Рис. 4.
Зависимости изменения энергии Гиббса реагентов реакции диссоциации малеиновой кислоты по первой ступени при переносе из воды в водный раствор диметилсульфоксида от состава водно-диметилсульфоксидного растворителя: 1 – [∆trG0(L2–) – ∆trG0(HL–)], 2 – ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2}}}}^{0}$ , 3 – ∆trG0(H+).
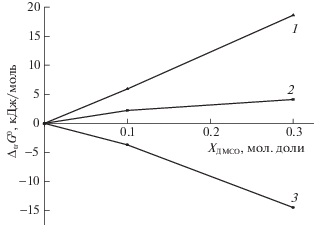
Рис. 5.
Зависимости изменения энергии Гиббса реагентов реакции диссоциации малеиновой кислоты по второй ступени при переносе из воды в водный раствор диметилсульфоксида от состава водно-диметилсульфоксидного растворителя: 1 – [∆trG0(L2–) – ∆trG0(HL–)], 2 – ${{\Delta }_{{{\text{tr}}}}}G_{{{\text{r2}}}}^{0}$ , 3 – ∆trG0(H+).
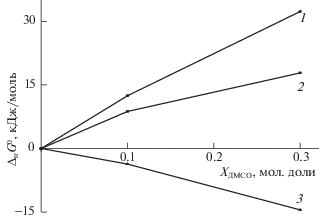
При исследовании диссоциации уксусной кислоты в водных растворах этанола и ДМСО, авторами работы [37] отмечалось, что основной вклад в уменьшение ее силы при увеличении содержания неводного компонента в растворе вносит дестабилизация ацетат-иона. В работе [38] авторы делают вывод, что изменения величин констант кислотной диссоциации глицина определяются влиянием растворителя на сольватацию всех участников кислотно-основного равновесия, а различия в характере зависимостей для pK1 и pK2 связаны с различным изменением сольватации протонированной и депротонированной форм глицина.
В табл. 6 представлены результаты расчетов энтальпий (${{\Delta }_{{\text{r}}}}H_{{298}}^{0}$) и энергий Гиббса (${{\Delta }_{{\text{r}}}}G_{{298}}^{0}$) депротонирования малеиновой кислоты при диссоциации по первой и второй ступени в газовой фазе и водном растворе (в рамках модели PCM). Несмотря на достаточную близость полученных величин, можно отметить некоторые закономерности. Депротонированные транс-формы Н2L, образованные при разрыве связи О4–Н, соответствующие минимуму на ППЭ, как и нейтральные изомеры, имеют плоское строение, в то время как при разрыве связи О1–Н плоская структура соответствует седловой точке первого порядка. В равновесной конформации НL– в последнем случае происходит разворот фрагмента О1–С1–О2 от плоскости молекулы на 39.0°, 45.7° в изомерах t1 и t2, соответственно. Причем, энергии депротонирования в обоих транс-изомерах выше в случае разрыва связи О4–Н. Интересно отметить, что депротонирование транс-изомеров по второй ступени приводит к стабилизации плоской структуры с симметрией CS. В случае цис-изомеров разрыв внутримолекулярной водородной связи (ВВС) несколько повышает энергию депротонирования. Кроме того, диссоциация по первой ступени, протекающая с разрывом ВВС приводит к изменениям в структуре молекулы: фрагмент ОСО, в котором произошел отрыв протона, разворачивается относительно плоскости молекулы на 93.0°. В равновесной конфигурации депротонированной цис-формы L2– один из фрагментов ОСО составляет единый плоский каркас с остальными атомами, в то время как другой фрагмент ОСО остается развернутым относительно плоскости молекулы на 94°.
Таблица 6.
Энтальпии и энергии Гиббса (ккал/моль) депротонирования малеиновой кислоты в газовой фазе и водном растворе по данным квантово-химических расчетов (B3LYP/cc-pVTZ)
| Изомер
(разрыв связи на первой ступени) |
Диссоциация по первой ступени | Диссоциация по второй ступени | ||
|---|---|---|---|---|
| ${{\Delta }_{{\text{r}}}}H_{{298}}^{0}$ | ${{\Delta }_{{\text{r}}}}G_{{298}}^{0}$ | ${{\Delta }_{{\text{r}}}}H_{{298}}^{0}$ | ${{\Delta }_{{\text{r}}}}G_{{298}}^{0}$ | |
| в газовой фазе | ||||
| с1(O1–H) | 313.8 | 306.9 | 454.6 | 445.2 |
| с1(O4–H) | 313.8 | 306.9 | 454.6 | 445.2 |
| с2(O1–H) | 335.6 | 328.1 | 432.8 | 424.8 |
| с2(O4–H) | 313.8 | 307.8 | 454.6 | 445.2 |
| t1(O1–H) | 333.6 | 325.7 | 422.6 | 415.5 |
| t1(O4–H) | 335.9 | 327.7 | 420.4 | 413.5 |
| t2(O1–H) | 332.9 | 325.0 | 422.6 | 415.5 |
| t2(O4–H) | 335.1 | 327.0 | 420.4 | 413.5 |
| в водном растворе | ||||
| с1(O1–H) | 272.6 | 265.3 | 308.2 | 299.6 |
| с1(O4–H) | 287.6 | 279.3 | 293.2 | 285.6 |
| с2(O1–H) | 283.5 | 275.9 | 295.0 | 287.5 |
| с2(O4–H) | 270.3 | 263.8 | 308.2 | 299.6 |
| t1(O1–H) | 283.3 | 275.7 | 293.0 | 285.1 |
| t1(O4–H) | 285.6 | 277.8 | 290.8 | 283.0 |
| t2(O1–H) | 283.2 | 275.6 | 292.8 | 285.0 |
| t2(O4–H) | 285.2 | 277.6 | 290.8 | 283.0 |
Общие закономерности в изменении структуры при депротонировании, а также в величинах энтальпий (${{\Delta }_{{\text{r}}}}H_{{298}}^{0}$) и энергий Гиббса (${{\Delta }_{{\text{r}}}}G_{{298}}^{0}$) сохраняются и при переходе в водный раствор. Можно отметить общую тенденцию к значительному уменьшению ${{\Delta }_{{\text{r}}}}H_{{298}}^{0}$ и ${{\Delta }_{{\text{r}}}}G_{{298}}^{0}$ в растворе.
Очевидно, что сумма энергий Гиббса сольватации ионов Н+ и L2– в растворителе во много раз превышает вклад от энергии сольватации Гиббса самой кислоты НL–, что значительно понижает энергетический барьер ее диссоциации. Поэтому энергия Гиббса газофазной диссоциации НL– в общем случае всегда больше, чем в растворе. В то же время, заметим, что применяемая здесь модель (PCM) учитывает только неспецифические типы взаимодействий “растворитель-растворенное вещество”, тогда как экспериментальные значения pKa зависят в значительной степени от специфической сольватации.
Исследование проведено в рамках государственного задания Министерства науки и высшего образования Российской Федерации (проект FZZW-2020-0009).
Список литературы
Amaro-Gahete J., Esquivel D., Ruiz J.R. et al. // Applied Catalysis A. General 2019. V. 585. P. 117190.
Bui N.-Q., Geantet C., Berhault G. //Applied Catalysis A: General. 2019. V. 572. P. 185.
Kim E. J., Haldar U., Lee H.-il // Polymer 2020. V. 186. P. 122040.
Voskoboynikova N., Orekhov P. S. et al. // Biochimica et Biophysica Acta (BBA) − Biomembranes. 2020. V. 1862. № 5. 183207.
Yamdagni R., McMahon T.B., Kebarle P. // J. Am. Chem. Soc. 1977. V. 99:7. P. 2222.
Koppel I.A., Burk P., Koppel I. et al. // J. Am. Chem. Soc. 2000. V. 122. № 21. P. 5114.
Shubin L., Schauer C.K., Pedersen L.G. // J. Chem. Phys. 2009. V. 131. P. 164107.
Koppel I.A., Taft R.W., Anvia F. et al. // J. Am. Chem. Soc. 1994. V. 116. № 7. P. 3047.
Fiedler P., Bohm S., Kulhanek J., Exner O. // Org. Biomol. Chem. 2006. V. 4. P. 2003.
Bohm S., Fiedler P., Exner O. // New J. Chem. 2004. V. 28. № 1. P. 67.
Wiberg K.B. // J. Org. Chem. 2002. V. 67. P. 4787.
Vianello R., Maksic Z.B. // Tetrahedron. 2006. V. 62. P. 3402.
Иванов С.Н., Гиричева Н.И., Нуркевич Т.В., Федоров М.С. // Журн. физ. химии. 2014. Т. 88. № 4. С. 647.
Koppel I.A., Taft R.W., Anvia F. et al. // J. Am. Chem. Soc. 1994. V. 116. № 7. P. 3047.
Тукумова Н.В., Усачева Т.Р., Чан Тхуан, Шарнин В.А. // Изв. вузов. Хим. хим. технология. 2011. Т. 54. № 4. С. 34.
Тукумова Н.В., Чан Тхи Зьеу Тхуан, Усачева Т.Р., Шарнин В.А. // Там же. 2012. Т. 55. № 9. С. 16.
Тукумова Н.В., Усачева Т.Р., Чан Тхи Зьеу Тхуан, Шарнин В.А. // Журн. физ. химии. 2014. Т. 88. № 10. С. 1512 .
Бородин В.А., Васильев В.П., Козловский Е.В. // Журн. неорган. химии. 1986. Т. 31. № 1. С. 10.
Daniele P.G., De Robertis A., De Stefano C. et al. // J. Chem. Soc., Dalton Trans. 1985. P. 2353.
Васильев В.П. Термодинамические свойства растворов электролитов / М.: Высш. школа, 1982. 320 c.
Лурье Ю.Ю. Справочник по аналитической химии. М.: Химия, 1989. 448 c.
Wooley E.M., Hurkot D.G., Hepler L.G. // J. Phys. Chem. 1970. V. 74. № 22. P. 3908.
Ferroni G., Galea J. // Ann. Chim. 1975. V. 10. № 1. P. 41.
Kalidas C., Hefter G., Marcus Y. // Chemical reviews. 2000. V. 100. № 3. P. 820.
Wells C.F. // J. Chem. Soc. Faraday Trans. 1979. V. 75. P. 53.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. // Gaussian 03, Revision B.03, Gaussian, Inc. 2003, Pittsburgh PA.
Bauschlicher C.W., Partridge H. // Chem. Phys. Lett. 1994. V. 231. P. 277.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Becke A.D. // Phys. Rev. A. 1998. V. 38. P. 3098.
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1998. V. 37. P. 785.
Vosko S.H., Wilk L., Nusair M. // Can. J. Phys. 1980. V. 58. P. 1200.
Dunning T. H. J.// J. Chem. Phys. 1989. V. 90. P. 1007.
Glendening E.D., Reed A.E., Carpenter J.E., Weinhold F. // QCPE Bull. 1990. V. 10. P. 58.
Zhurko G.A., Zhurko D.A. ChemCraft version 1.6 (build 312) ed. http://www.chemcraftprog.com/index.html
Foresman J.B., Keith T.A., Wiberg K.B. et al. // J. Chem. Phys. 1996. V. 100. № 40. P. 16098.
Смирнова Н.А. Методы статистической термодинамики в физической химии. М.: Высш. школа, 1973. 480 с.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Журн. физ. химии. 1997. Т. 71. № 8. Р. 1371.
Исаева В.А., Леденков С.Ф., Шарнин В.А., Шорманов В.А. // Там же. 1993. Т. 67. № 11. С. 2202.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии