

Журнал физической химии, 2021, T. 95, № 7, стр. 1078-1088
Молекулярно-слоевое осаждение и пиролиз пленок полиамида на Si(111) с образованием β-SiC
Р. Р. Амашаев a, И. М. Абдулагатов a, *, М. Х. Рабаданов a, А. И. Абдулагатов a
a Дагестанский государственный университет
367000 Махачкала, Россия
* E-mail: ilmutdina@gmail.com
Поступила в редакцию 01.09.2020
После доработки 20.10.2020
Принята к публикации 21.10.2020
Аннотация
Проведено молекулярно-слоевое осаждение (МСО) тонких пленок полиамида с использованием прекурсоров 1,3,5-бензолтрикарбонилтрихлорида (тримезоилхлорида, TMХ) и 1,2-этилендиамина (ЭДА) при температуре 120°С; постоянная роста пленки при данной температуре составляла 1.85 нм/цикл. Для определения характера роста пленки использованы in situ кварцевые пьезоэлектрические микровесы (КПМ). Установлен линейный рост пленки с увеличением количества МСО-циклов. Пиролиз МСО полиамидных пленок на Si(111) проведен при температурах 1100 или 1300°С и при давлении 10–7 Торр. В результате твердофазной реакции Si и C при 1300°С на поверхности получены тонкие гетероэпитаксиальные пленки кубического β-SiC (3C–SiC). Различные спектроскопические методы высокого разрешения использованы для определения элементного состава и кристаллической структуры полученных органических и керамических пленок.
Благодаря уникальной комбинации механических, электрических, оптических и химических свойств карбид кремния (SiC) является одним из важных индустриальных материалов [1]. Различные политипы SiC служат эффективными квантовыми излучателями как с оптической накачкой, так и с электрическим приводом в видимом и телекоммуникационном диапазонах волн [2, 3]. Карбид кремния SiC на кремнии может быть интегрирован в комплементарные структуры металл–оксид–полупроводник (КМОП) для производства продвинутых полевых [4, 5] и диффузионных транзисторов [6], а также оптических волноводов и кольцевых резонаторов [7]. С недавнего времени SiC считается перспективным материалом для изготовления электронной базы квантовых компьютеров [8, 9]. Кроме этого, SiC может быть использован для получения другого технологически важного материала – графена [10, 11].
Дальнейшее развитие технологии SiC в приложениях микроэлектромеханических систем (МЭМС) требует выращивания монокристаллического или поликристаллического 3C–SiC на подложках большой площади, которые совместимы с экономичными высокопроизводительными процессами серийного производства, используемыми в микросхемах кремния и производстве интегральных схем [12]. Традиционно пленки карбида кремния осаждаются из газовой фазы методами магнетронного распыления [13], химического осаждения из газовой фазы (ХОГФ) [14], комбинацией различных плазмохимических методов и ХОГФ [15–17], карбонизации [18–20] и т.д. В другом способе, полиимидные пленки, полученные жидкофазным методом Ленгмюра–Блоджетт на кремниевой подложке, использовали для синтеза пленок SiC путем термической обработки в инертной атмосфере или вакууме [21–24]. Однако при использовании этих методов сталкиваются с проблемами, касающимися равномерности осаждения и состава пленок [25, 26], которые в конечном счете влияют на свойства полученных пленок [27].
В данной работе описан новый способ получения равномерных тонких пленок SiC [заявка на патент находится на рассмотрении] путем термообработки в вакууме или инертной среде тонких МСО-пленок полиамида на кремниевой подложке. Молекулярно-слоевое осаждение (МСО) – это метод осаждения из паровой фазы органических или гибридных органо-неорганических тонких пленок за счет термически стимулированных поверхностных реакций органических или комбинации органических и неорганических прекурсоров [28]. Данный метод базируется на методе “молекулярного наслаивания”, известного также как атомно-слоевое осаждение (АСО), разработанного в 60-x годах прошлого столетия научной школой член-корреспондента АН СССР В.Б. Алесковского [29]. Основное преимущество МСО-технологии над другими методами осаждения состоит в обеспечении точного контроля толщины, состава и конформности осаждаемых пленок, как на наноструктурированных плоских подложках, так и на дисперсных материалах [30]. Данный способ получения тонких пленок не требует дорогостоящего оборудования и может быть относительно легко приспособлен для производства партиями или конвейерным способом. На данный момент разработаны процессы МСО полиимидов [31], полиамидов [32, 33], полимочевины [34], полиазометинов [35], полиэфиров [36] и т.д. Гибридные органо-неорганические алкоксидные МСО-пленки могут быть использованы для получения тонких оксид-графитных керамических покрытий [37, 38] и графитных пленок путем пиролиза полимерных МСО-пленок [39]. Использование полимерной пленки полиамида, нанесенной на кремниевую подложку методом МСО, в качестве прекурсора для синтеза пленок SiC, сделает возможным точный контроль толщины и однородности пленки SiC благодаря контролю параметров исходной пленки. МСО-пленки полиамида в данной работе осаждали с использованием тримезоилхлорида и 1,2-этилендиамина с последующим пиролизом МСО-пленки при 1100 или 1300°C в вакууме для получения пленки карбида кремния.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
МСО-пленки полиамида получали на оборудовании компании АСО НаноТех (г. Махачкала, Россия). Установка снабжена вакуумной камерой с горячими стенками, куда натекает вязкий поток инертного газа. Азот ультравысокой чистоты (УВЧ, 99.9999%) использовали в качестве газа- носителя. В процессе МСО давление в реакторе поддерживали на уровне ~1.0 Торр. Для осаждения использовали 1,3,5-бензолтрикарбонил трихлорид (тримезоилхлорид (TMХ), CAS номер 4422-95-1, 98%, Sigma-Aldrich) и 1,2-этилендиамин (ЭДА, CAS номер 107-15-3, 99.5%, Sigma-Aldrich). Прекурсоры загружали в контейнеры для дозирования в перчаточном боксе в атмосфере азота. Перед началом процесса все реагенты дегазировали.
В качестве подложек для осаждения МСО-пленок использовали полированные с одной стороны подложки Si(111) размером 1 × 1 см. Предварительно слой естественного оксида на поверхности Si травили 10%-ным водным раствором HF в течение 10 мин, после чего полоскали в потоке дистиллированной воды. Естественный слой оксида удаляли для улучшения нуклеации и роста пленки карбида в процессе пиролиза. Для доставки паров ТМХ в реакционную зону температуру контейнера с реагентом и линию подачи нагревали до 115°С.
Перед осаждением подложки помещали в реактор, где выдерживали в течение 1 ч для дегазации. Временные параметры одного МСО-цикла ТMХ–ЭДА были 1/30/1/30, где 1 с – время напуска ТMХ или ЭДА, 30 с – продолжительность продувки азотом после напуска прекурсоров. Толщину осаждаемой пленки регулировали количеством МСО-циклов. Парциальное давление ТМХ поддерживали на уровне ~0.05 Торр, а ЭДА – ~0.12 Торр.
Мониторинг процесса осаждения проводили в режиме реального времени (in situ) методом кварцевого пьезоэлектрического микровзвешивания (КПМ) с использованием Inficon STM-2 разрешением ~0.37 нг/см2 и резонансной частотой AT-среза кристалла 6 МГц.
Спектроскопию комбинацинонного рассеяния (СКР) использовали для определения структуры полученных пленок до и после пиролиза. Для этого использовали спектрометр NTEGRA SPECTRA в диапазоне длин волн 1900–100 см–1 с длиной волны возбуждения 532 нм.
Сканирующий электронный микроскоп (СЭМ) использовали для снятия изображения поверхности образцов и их поперечного среза. СЭМ-изображения снимали на оборудовании Sem-Tescan Mira, X-Max EDS 50. Опцию энергодисперсной рентгеновской спектроскопии (ЭДРС) с энергией электронов 5 кэВ использовали для определения элементного состава пленок.
Дифракцию рентгеновских лучей (ДРЛ) и дифракцию быстрых электронов на отражение (ДБЭО) использовали для определения кристалличности структуры пленки после пиролиза. Анализ ДБЭО проводили на оборудовании ЭД-75 с напряжением 71 кВ. Анализ ДРЛ проводили на дифрактометре Empyrian II, оснащенном медной трубкой (CuKα, λ = 1.5405 Å) и линейным детектором Pixelid.
Пиролиз МСО пленок проводили в вакуумном колпаке при давлении 10–7 Торр с использованием криогенного насоса. В качестве нагревательного элемента, на котором были размещены образцы пленок, использовали очищенную тонкую молибденовую пластину. Перед процессом пиролиза внутренние стенки системы дегазировали вакуумной лампой накаливания в течение 1 ч при давлении 10–7 Торр. Во время пиролиза в начале температуру довели до 450°С со скоростью нагрева 5 К/мин и временем задержки 30 мин. Затем до 1100 и 1300°С со скоростью 10 К/мин и временем задержки 1 ч для каждой из пиковых температур. Задержку при 450°С проводили для избежания резкого выделения летучих компонентов при более высоких температурах [40], что могло привести к неравномерности распределения углерода после пиролиза. Охлаждение происходило естественным путем.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
МСО полиамида: данные КПМ
Алифатические полимеры, в частности алифатические полиамиды, почти полностью разлагаются на летучие компоненты в инертной атмосфере и вакууме [41], в то время как пиролиз ароматических полимеров, например. ароматических полиамидов или полиимидов [42, 43], дает углеродный остаток более 50% от начальной массы в зависимости от степени ароматичности [40]. В связи с этим нами была разработана химия МСО для получения ароматического полиамида. Предлагаемая схема осаждения полиамида, состоящая из (А)–(Б) поверхностных реакций, представлена на рис. 1. В первой полуреакции (А) представлено взаимодействие хлорангидридных групп ТМХ с поверхностными –NH2-группами ЭДА с образованием амидных связей и молекул HCl как продуктов поликонденсации. В следующей МСО-полуреакции (Б) напуск ЭДА ведет к реакции первичного амина –NH2 c поверхностными хлорангидридными функциональными группами, в результате которых регенерируются начальные функциональные группы. Как показано на рисунке, гомофункциональная молекула ЭДА, может реагировать как с одной, так и двумя функциональными –NH2-группами одновременно [33]. В результате также образуются амидные связи и HCl. Варьирование количества МСО-циклов позволяет прецизионно контролировать толщину пленки полиамида.
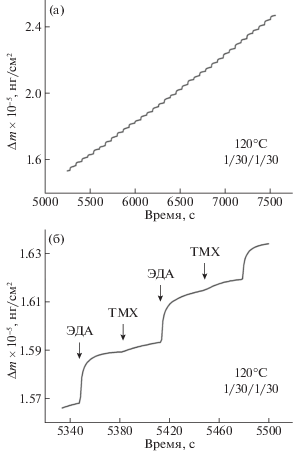
На рис. 2 показаны КПМ-данные прироста массы в процессе МСО при температуре осаждения 120°С и временными параметрами цикла 1/30/1/30. На рис. 2а представлен сигнал КПМ в течение ~35 МСО-циклов ТМХ–ЭДА, показывающий линейный рост пленки в зависимости от времени (количества циклов). На рис. 2б показан сигнал КПМ для двух МСО-циклов, где виден прирост массы в результате индивидуального напуска реагентов. Отсюда прирост массы после напуска ЭДА в течение 1 с составил 2094 нг/см2, а в случае ТМХ – 397 нг/см2 с общим приростом массы для одного МСО-цикла 2491 нг/см2.
Рис. 2.
КПМ-сигнал прироста массы в процессе МСО ТМХ–ЭДА при 120°C для 35 (а) и двух (б) МСО-циклов.
Относительно высокое значение прироста массы за цикл для системы МСО TMХ–ЭДА, вероятно, связано с диффузией ЭДА и в меньшей степени ТМХ в пленку. Это можно объяснить уменьшением плотности сшивания полимера из-за относительно высокой температуры процесса МСО, вследствие чего полимер приобретает большую абсорбционную способность. Низкая плотность сшивания может быть результатом теплового движения гибких полимерных цепей, между которыми образуются небольшие пространства, в которые могут проникать молекулы прекурсоров [44]. Абсорбционные процессы косвенно подтверждаются КПМ-данными. Полученные данные КПМ не соответствуют предложенной схеме осаждения (рис. 1). Согласно схеме, прирост массы при напуске ТМХ должен быть в ~4.8 раза больше по сравнению с наблюдаемым в случае ЭДА. Значительно больший прирост массы после дозирования ЭДА может быть связан с меньшим размером молекулы ЭДА (3.75 Å) по сравнению с более объемной молекулы TMХ (7.26 Å), вследствие чего ЭДА, испытывая в меньшей степени влияние стерических эффектов, легче поглощается пленкой. Двойные реакции ЭДА с поверхностью также могут вносить вклад в меньший прирост массы при напуске ТМХ вследствие уменьшения количества поверхностных функциональных групп. Аналогичный пример, когда значительный прирост массы наблюдался во время МСО полимера, описывается в работе [45], где процесс МСО представляет собой трехступенчатый AБВ-цикл с использованием циклического азасилана (АС), малеинового ангидрида (MA) и H2O для осаждения органо-неорганических пленок. После 50 АБВ-циклов при 100°C прирост массы стабилизировался на уровне 1510 нг/(см2 цикл).
В то же время значительный прирост массы может происходить также за счет физической конденсации реагентов и/или процессов по типу ХОГФ. Эксперимент показал, что прекращение дозирования прекурсора ТМХ в МСО-цикле приводило к деградации линейного прироста массы, что свидетельствует о минимальном вкладе конденсационных процессов и о обеспечении роста за счет поверхностных реакций.
Анализ МСО пленок: данные СКР
Образцы полиамида на Si(111) до и после пиролиза при 1100 и 1300°С анализировали с применением спектроскопии комбинационного рассеяния (СКР). МСО-пленку осаждали на Si(111) при 120°С 300 циклами ТМХ–ЭДА с временными параметрами 1/30/1/30. Анализ полученной полиамидной пленки в диапазоне 1900–100 см–1 показал следующие основные пики: 885, 995, 1292, 1338, 1364, 1432, 1552, 1594 и 1638 см–1 (рис. 3). Схожий спектр КР наблюдали для пленки полиамида толщиной 200 нм, полученной методом МСО с использованием тримезоилхлорида и м-фенилендиамина [46], а также в работе, где полиамидная мембрана была получена из жидкой фазы [47]. Пик при 995 см–1 является самым высоким. СКР-пик вблизи 1000 см-1 характерен для всех производных бензола и связан c деформационными колебаниями С–Н в бензольном кольце [48]. Пики 1292 и 1432 см–1 связаны с синфазными кручениями –CH2-групп [49]. Пик 1552 см–1 относится к колебаниям C–N–H при изгибе плоскости. Наличие пика при 1338 см–1 может быть связано с валентными колебаниями $ > {\kern 1pt} {\text{C}}{\kern 1pt} --{\kern 1pt} {\text{N}}{\kern 1pt} + {\kern 1pt} \bullet $ делокализованных поляронных носителей заряда [50]. Пик 1638 см–1 относится к валентным колебаниям амидной группы –С=O (полоса амида I). Слабый пик при 1726 см–1 относится к асимметричным валентым колебаниям карбонильной группы –С=O в составе производных ароматических соединений и в сложноэфирных группировках [51]. Второй по величине пик полиамида при 1594 см–1 относится к связи С=С в бензольном кольце. Пик при 885 см–1 может свидетельствовать о наличии валентных колебаний связей С-фенил и C–Cl [51].
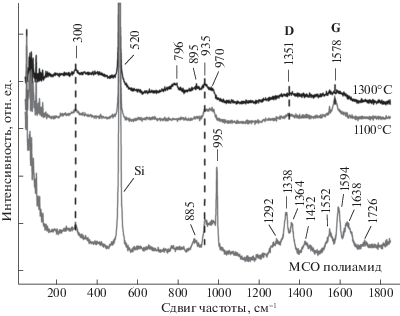
Для пленки, пиролизованной при 1100°C, спектры КР показали широкий пик при 1351 см–1 и более отчетливый пик при 1578 см–1. Эти пики наблюдаются в спектрах аморфных углеродных материалов, содержащих нанокристаллические графитовые домены, и обозначаются как D (1351 см–1) и G (1578 см–1). Пики D и G характерны для углерода в sp2-гибридизации. Пик D связан с колебательными модами sp2-атомов углерода в неупорядоченной углеродистой структуре [52, 53]. Пик G обусловлен валентными колебаниями связи C–C sp2 углерода как в ароматических кольцах, так и в углеродных цепях и отображает степень кристаллизации [54]. Более высокая интенсивность пика G по сравнению с пиком D, вероятно, связана с высокой степенью кристаллизации образовавшейся графитовой пленки [55].
При повышении температуры пиролиза до 1300°С появляются пики при 796, 895, 935 и 970 см–1. Пик 796 см–1 обусловлен поперечными (TO) оптическими колебаниями связей Si–C, а слабый пик при 970 см–1 может быть связан с продольными (LO) оптическими колебаниями Si–C. Пик при 895 см–1 относится к А1 SiC [56]. Позиции пиков при 796 см–1 и 970 см–1 соответствуют кубическому карбиду кремния (3С–SiC) [57]. Снижение интенсивности пиков D и G (1351 и 1578 см–1 соответственно) можно объяснить твердофазной реакцией свободного углерода с кремниевой подложкой при повышении температуры от 1100 до 1300°С. Пики при 300, 520 и 935 см–1 относятся к кремниевой подложке.
Отсутствие LO- и TO-пиков SiC при 1100°С не означает полного отсутствия SiC. Согласно данным [58–60], образование тонкой пленки SiC возможно даже при температурах 800–950°C. Это может свидетельствовать о малой толщине образовавшейся пленки SiC и низкой чувствительности инструмента. Возможно, необходимо более длительное время термообработки (>1 ч) для полной реакции свободного углерода.
Морфология и состав пленок: СЭМ- и ЭДРС-анализ
На рис. 4а приведено СЭМ-изображение поперечного скола образца МСО полиамидной пленки на Si(111), полученной 300 циклами TMХ–ЭДА при 120°С. Согласно данным рис. 4, толщина полученной пленки составила ~556.6 нм. Соответственно постоянная роста МСО-пленки составляет 1.85 нм/цикл. Согласно расчетам [61], длина структурного звена полиамида, полученного с использованием TMХ и ЭДА, должна составлять ~1.3 нм. Значение постоянной роста выше длины структурного звена, предположительно, вследствие диффузии реагентов в процессе осаждения и возможных побочных процессов по типу ХОГФ.
Рис. 4.
СЭМ-изображение поперечного скола поламидной МСО-пленки на Si(111): а – до термообработки, б – после пиролиза при 1100°C, в – после пиролиза при 1300°C; г – иллюстрация пустотного дефекта под пленкой, пиролизованной при 1300°C.

На рис. 4б представлено изображение образца МСО-пленки на Si(111) после пиролиза при 1100°C. После пиролиза наблюдалось уменьшение толщины пленки до 109 нм, что является ~5-кратным снижением толщины. Массовая доля углерода бензольного кольца в структурной единице ТМХ–ЭДА полиамида составляет 29.3%, т.е. почти треть от общей массы. В соответствии с работой [40], где, как сообщалось, бензольные кольца полимеров служат основным источником остаточного углерода после пиролиза, снижение толщины полиамидной пленки после пиролиза должно быть в 3 и более раз. Согласно СКР-данным (рис. 3) пленка представляет собой графитизированный углерод.
На рис. 4в показан поперечный скол пленки после пиролиза при 1300°С. На кремниевой подложке видны два гетерогенных слоя. Верхний слой имеет толщину ~42.3 нм, а нижний – ~43.8 нм. На основании СКР-данных можно предположить, что верхний слой состоит из непрореагировавшего графитизированного углерода, а нижний – из 3С–SiC. С другой стороны, данные два слоя могут быть слоями поликристаллического и монокристаллического β-SiC (3С–SiC), формирование которых наблюдалось в работе [22], где пленки SiC были получены карбонизацией полиимидных пленок Ленгмюра–Блоджетт на подложке Si(111), а также в работе [62], где пленки SiC на кремнии были получены методом ХОГФ.
СЭМ-анализ образцов, полученных при 1300°С, выявил также образование пустот под пленкой SiC, которые предположительно являются дефектами, вызванными диффузией Si из подложки в реакционную поверхность [63]. Одна из таких пустот изображена на рис. 4д. Схожую картину наблюдали, к примеру, при пиролизе полиимидных пленок Ленгмюра–Блоджетт при 1200°С на подложке кремния [22].
На рис. 5 представлены СЭМ-изображения поверхности МСО-пленки до и после пиролиза при 1100 и 1300°С. На рис. 5а показана поверхность полиамидной МСО-пленки толщиной 556.6 нм, полученной при 120°С до пиролиза. СЭМ-изображение выявило характерную зернистую структуру пленки. Аналогичная морфология поверхности пленок и мембран ароматических полиамидов на основе тримезоилхлорида была отмечена в работах [64–66]. На рис. 5б показана поверхность пленки после пиролиза полиамидной пленки при 1100°C. Полученная при данной температуре пленка была относительно гладкая и без дефектов. На рис. 5в представлено изображение поверхности образца после пиролиза при 1300°С, где наблюдали образование ям на поверхности пленки. Аналогичные дефекты в системе SiC/Si в диапазоне температур от 1100 до 1300°С наблюдали в работах [67–69]. Более детальное изображение поверхности пленки после пиролиза при 1300°С, представленное на рис. 5г, свидетельствует о формировании шероховатого и гранульного рельефа поверхности, характерного для кристаллических пленок с большим размером зерен.
Рис. 5.
СЭМ-изображения поверхности: а – полиамидной МСО-пленки толщиной 556.6 нм, б – после пиролиза при 1100°C, в – после пиролиза при 1300°С, г – 10-кратное увеличение для пленки при 1300°C.

Элементный состав образцов до и после пиролиза определяли с помощью ЭДРС с энергией электронов 5 кэВ. Получен следующий элементный состав полиамидной МСО-пленки: C – 61.31 ат. %, N – 19.54 ат. %, O – 15.59 ат. %, Cl – 3.34 ат. % и Si – 0.22 ат. %. Присутствие незначительного количества кремния связано с высоким разрешением ЭДРС по глубине. Наличие примеси Cl в пленке может указывать на неполноту поверхностных реакций или неполную десорбцию HCl на этапе продувки. Рассчитанный состав структурного звена полиамида: C – 66.6 ат. %, N – 16.6 ат. % и O – 16.6 ат. %. Таким образом, определенный по ЭДРС элементный состав МСО полиамидной пленки хорошо согласуется с расчетным составом структурного звена TMХ–ЭДА-полиамида. Из атомарного отношения N/O можно оценить степень сшивания полиамида, причем чем ближе отношение N/O к 1, тем выше степень сшивания полиамида. Отношение N/O = =1.25 указывает на высокую степень сшивания полиамидной МСО-пленки.
ЭДРС-анализ пленки после пиролиза при 1100°С показал следующий элементный состав: C – 74.02 ат. %, N – 3.65 ат. %, O – 1.27 ат. % и Si – 21.06 ат. %. Как видно, пиролиз приводит к значительному увеличению содержания углерода и кремния. Снижение концентрации N и O после пиролиза обусловлено механизмом термического разложения ароматического полиамида, которое начинается с расщепления амидной связи и образования CO2, CO, HCN, CH4, H2, H2O [40]. После пиролиза содержание хлора стало ниже чувствительности инструмента. В случае, если примеси Cl присутствуют в МСО-пленке в форме непрореагировавших хлорангидридных групп, атомы хлора могут реагировать с водородом органических компонентов при относительно низких температурах с образованием HCl и, тем самым, стимулировать карбонизацию [70]. После пиролиза при 1300°C элементный состав пленки составил: C – 50.03 ат. %, O – 0.59 ат. % и Si – 49.38 ат. %. В пленке отсутствовали примеси азота. Примеси кислорода, вероятно, присутствуют в форме карбоксильных и гидроксильных групп. Элементы C и Si в данном образце содержатся почти в равных пропорциях, что может свидетельствовать о том, что два обнаруженных слоя, представленные на рис. 4в и 4г, являются наноламинатом поликристаллического SiC (верхний слой) и монокристаллического 3С–SiC (нижний слой) с общей толщиной 86.1 нм. Для минимизирования влияния подложки на элементный состав, ЭДРС-анализ проводился при относительно низкой энергии электронов 5 кэВ.
Кристаллическая структура пленок: ДБЭО- и ДРЛ-анализ
Методами дифракции быстрых электронов на отражение (ДБЭО) и дифракции рентгеновских лучей (ДРЛ) определяли кристаллическую структуру пиролизованных образцов пленок. На рис. 6 представлена дифракционная картина поверхности образцов, полученных пиролизом при 1300°С. На рис. 6а представлено ДБЭО-изображение для образца пленки после пиролиза при 1300°С в течение 1 ч при пиковой температуре. Наличие колец на ДБЭО-изображении свидетельствует о поликристаллической природе поверхностной пленки. Схожий результат наблюдали в других работах, где пленки SiC были получены пиролизом при 1300°С [71, 72]. Появление данной картины связывали с формированием хаотично ориентированных микрокристаллов 3C–SiC. С целью получения дифракционной картины, характерной для упорядоченной структуры 3C–SiC, был проведен вторичный пиролиз того же образца при 1300°С и давлении 10–7 Торр в течение 1 ч. Исследование образцов пленок после вторичного пиролиза позволило обнаружить ДБЭО-картину, соответствующую 3C–SiC на кремнии по азимуту 〈110 〉 (рис. 6б)) [59, 73–75]. Данная ДБЭО-картина может указывать на двухосновную текстуру образовавшихся пленок, где преимущественно эпитаксиально ориентированные кристаллиты достигают приповерхностную область [76]. Наблюдаемая ДБЭО-картина имеет четкие, яркие рефлексы и низкий уровень фона, что свидетельствует о высоком качестве полученных пленок 3C–SiC.
Рис. 6.
ДБЭО-изображения, полученные пиролизом при 1300°C: а – поверхность поликристаллического SiC до вторичного пиролиза, б – в азимуте 〈110 〉 после вторичного пиролиза при 1300°С.

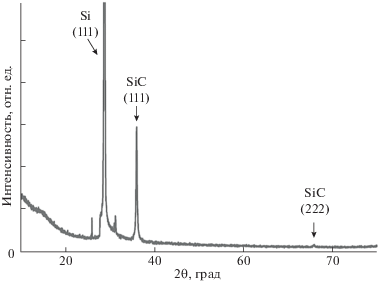
ДБЭО-анализ предоставляет информацию о кристалличности верхних монослоев пленки, в то время как ДРЛ дает информацию об объемной кристалличности. ДРЛ-дифрактограмма дважды пиролизованного при 1300°С образца пленки представлена на рис. 7. Пики при 2θ = 35.8° и 75.8°, согласно дифракционной карте ICSD (№ 98-002-4217), соответствуют дифракционным плоскостям 〈111 〉 и 〈222〉 3C–SiC. Из ICSD № 98-002-4217 интенсивности ДРЛ-пиков для 3С–SiC приходятся как 100% (2θ = 35.8°) и 4.6% (2θ = = 75.8°). Интенсивности пиков для пленки 3C–SiC, полученной пиролизом МСО-пленки, составили 100 и 6% соответственно. Пик при 25.9° можно отнести к графитоподобным структурам [77]. Значение полной ширины на уровне половинной амплитуды (FWHM) для пика при 35.8° составило 0.41°. В работе [78] для пленки 3C–SiC толщиной 83 нм значение FWHM составляло 0.4° ± ± 0.012° (2θ = 35.75°), что близко к значению для пленки 3C–SiC, полученной в данной работе. ДРЛ-данные подтверждают формирование гетероэпитаксиальной пленки 3C–SiC путем пиролиза полиамидной МСО-пленки на Si(111) при 1300°С.
Таким образом, описан процесс получения тонких пленок 3C–SiC путем пиролиза МСО-пленок ароматического полиамида на Si(111). КПМ-данные мониторинга роста полиамидной МСО-пленки показали линейность роста пленки с увеличением количества циклов. Постоянная роста МСО-пленки при 120°С составила ~1.85 нм/цикл. Спектр КР пиролизованного в течение 1 ч при 1300°C образца показал пики, характерные для кристаллического 3С–SiC. СЭМ-изображения того же образца показали формирование двухслойной гетероструктуры. ДБЭО-анализ показал поликристаллическую природу верхнего слоя. ЭДРС-анализ указал на равное атомарное соотношение C и Si в данном образце. После вторичного пиролиза ДБЭО показала дифракционную, относящуюся к монокристаллическому 3С–SiC. ДРЛ-анализ показал два пика, характерных для 3C–SiC после вторичного пиролиза. Помимо этого, на границе SiC/Si и на поверхности пленки SiC были обнаружены дефектные поры, что свидетельствует о необходимости оптимизации режимов пиролиза. Дальнейшие исследования равномерности и конформности синтеза пленок карбида кремния будут продолжены в следующих работах.
Авторы благодарят А.М. Исмаилова (кафедра физической электроники Дагестанского государственного университета) за помощь в получении ДБЭО-снимков и некоторую техническую помощь.
Данная работа выполнена при финансовой поддержке РФФИ в рамках научного проекта № 19-33-90045 (Р.Р. Амашаев) и частичной финансовой поддержке Правительства Российской Федерации в рамках гранта № ФЗНЗ-2020-0002 (И.М. Абдулагатов).
Список литературы
Saddow S.E., Agarwal A. Advances in Silicon Carbide Processing and Applications. Artech House; 2004.
Lukin D.M., Dory C., Guidry M.A. et al. // Nature Photonics. 2020. V. 14. № 5. P. 330.
Fuchs F., Stender B., Trupke M. et al. // Nature Communications. 2015. V. 6. № 1. P. 7578.
Duan B., Yang X., Lv J., Yang Y. // IEEE Transactions on Electron Devices. 2018. V. 65. № 8. P. 3388.
Jianwei W., Capano M.A., Melloch M.R., Cooper J.A. // IEEE Electron Device Letters. 2002. V. 23. № 8. P. 482.
Lotfi S., Li L.G., Vallin O. et al. // Solid State Electronics. 2012. V. 70. № P. 14. https://doi.org/10.1016/j.sse.2011.11.019
Powell K., Shams-Ansari A., Desai S. et al. // Opt Express. 2020. V. 28. № 4. P. 4938.
Weber J.R., Koehl W.F., Varley J.B. et al. // Journal of Applied Physics. 2011. V. 109. № 10. P. 102417.
Choudhary R., Biswas R., Pan B., Paudyal D. // MRS Advances. 2019. V. 4. № 40. P. 2217.
Presser V., Heon M., Gogotsi Y. // Advanced Functional Materials. 2011. V. 21. № 5. P. 810.
Hass J., de Heer W.A., Conrad E.H. // Journal of Physics: Condensed Matter. 2008. V. 20. № 32. P. 323202.
Zorman C.A., Rajgopal S., Fu X.A. et al. // Electrochem Solid State Lett. 2002. V. 5. № 10. P. G99.
Ledermann N., Muralt P., Xantopoulos N., Tellenbach J.-M. // Surface and Coatings Technology. 2000. V. 125. № 1–3. P. 246.
Brütsch R. // Thin Solid Films 1985. V. 126. № 3–4. P. 313.
Tong L., Mehregany M., Tang W.C., editor Amorphous Silicon Carbide Films by Plasma-Enhanced Chemical Vapor Deposition. IEEE Micro Electro Mechanical Systems; 1993; Fort Lauderdale, FL, USA.
Loboda M.J., Seifferly J.A., Dall F.C. // Journal of Vacuum Science & Technology A. 1994. V. 12. № 1. P. 90.
Cheng Q.J., Xu S.Y., Long J.D., Ostrikov K. // Chemical Vapor Deposition. 2007. V. 13. № 10. P. 561.
Severino A., D’Arrigo G., Bongiorno C. et al. // J. Appl. Phys. 2007. V. 102. № 2. P. 023518.
Watanabe Y., Horikawa T., Kamimura K. // Japan. J. Appl. Phys.2014. V. 53. № 4. P. 045601.
Molina S.I., Morales F.M., Araújo D. // Materials Science and Engineering: B. 2001. V. 80. № 1. P. 342.
Yang B., Zhou Y., Cai W. et al. // Appl. Phys. Lett. 1994. V. 64. № 11. P. 1445.
Luchinin V., Goloudina S., Pasyuta V. et al. // Japan. J. Appl. Phys. 2017. V. 56. № 6. P. 06GH08.
Pan H., Guo H., Lu E. et al. // J. Electron Spectroscopy and Related Phenomena 1999. V. 101. P. 685. https://doi.org/10.1016/S0368-2048(98)00390-9
Jin B., He P., Sheng Y., Yang B. // J. Physics and Chemistry of Solids. 2003. V. 64. № 2. P. 339.
Leal G., Campos T., Sobrinho A. et al. // Materials Research. 2014. V. 17. № 2. P. 472.
DeAnna R.G., Fleischman A.J., Zorman C.A., and Mehregany M. // J. Chem Vap Deposition 6. 1998. V. 6. № 4. P. 280.
Carballo J.M. Residual Stress Analysis in 3C–SiC Thin Films by Substrate Curvature Method: Theses and dissertations. University of South Florida, 2010.
George S.M., Yoon B., Dameron A.A. // Acc Chem Res. 2009. V. 42. № 4. P. 498.
Malygin A.A., Drozd V.E., Malkov A.A., Smirnov V.M. // Chemical Vapor Deposition. 2015. V. 21. № 10-11-12. P. 216.
George S.M. // Chem. Rev. 2010. V. 110. № 1. P. 111.
Yoshimura T., Tatsuura S., Sotoyama W. // Appl. Phys. Lett. 1991. V. 59. № 4. P. 482.
Du Y., George S.M. // J. Phys. Chem. C. 2007. V. 111. № 24. P. 8509.
Adamczyk N.M., Dameron A.A., George S.M. // Langmuir. 2008. V. 24. № 5. P. 2081.
Loscutoff P., Zhou H., Clendenning S., Bent S. // ACS nano. 2009. V. 4. № 1. P. 331.
Yoshimura T., Tatsuura S., Sotoyama W. et al. // Appl. Phys.Lett.1992. V. 60. № 3. P. 268.
Ivanova T.V., Maydannik P.S., Cameron D.C. // J. Vacuum Science & Technology A. 2012. V. 30. № 1. P. 01A121.
Abdulagatov A., Terauds K., Travis J. et al. // J. Phys. Chem. C.2013. V. 117. № 34. P. 17442.
DuMont J.W., George S.M. // Ibid. 2015. V. 119. № 26. P. 14603.
Yang P., Wang G., Gao Z. et al. // Materials (Basel). 2013. V. 6. № 12. P. 5602.
Ehlers G.F.L., Fisch K.R., Powell W.R. // J. Polymer Science Part A-1: Polymer Chemistry. 1970. V. 8. № 12. P. 3511.
Herrera M., Matuschek G., Kettrup A. // J. Thermal Analysis and Calorimetry. 2000. V. 59. № 1–2. P. 385.
Hatori H., Yamada Y., Shiraishi M. et al. // Carbon. 1996. V. 34. № 2. P. 201.
Xie W., Pan W.-P., Chuang K. // J. Thermal Analysis and Calorimetry. 2001. V. 64. № 2. P. 477.
Krongauz V. // Ibid. 2010. V. 102. № 2. P. 435.
Ju L. New Chemistries and Processing in Atomic and Molecular Layer Deposition of Inorganic and Hybrid Organic-Inorganic Thin Films: Theses and Dissertations. Lehigh University, 2018.
Higgs D.J., DuMont J.W., Sharma K., George S.M. // J. Vacuum Science & Technology A. 2018. V. 36. № 1. P. 01A117.
Kim H.J., Choi K., Baek Y. et al. // ACS Appl Mater Interfaces. 2014. V. 6. № 4. P. 2819.
Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra. Chapter 9 – Benzene and Its Derivatives / Ed. by R.A.Nyquist, San Diego: Academic Press, 2001. P. 351.
Pud A.A., Fatyeyeva K.Y., Bardeau J.F. et al. // J. Macromolecular Science, Part A. 2007. V. 44. № 2. P. 183.
Furukawa Y., Ueda F., Hyodo Y. et al. // Macromolecules. 1988. V. 21. № 5. P. 1297.
Socrates G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts. 3 ed: Amer. Chem. Soc., 2002. 366 p.
Ferrari A.C., Robertson J. // Phys. Rev. B. 2001. V. 64. № 7. P. 075414.
Maultzsch J., Reich S., Thomsen C. // Ibid. 2004. V. 70. № 15. P. 155403.
Ferrari A.C. // Ibid.2000. V. 61. № 20. P. 14095.
Ohnishi T., Murase I., Noguchi T., Hirooka M. // Synthetic Metals. 1987. V. 18. № 1. P. 497.
Burton J.C., Sun L., Pophristic M. et al. // J. Appl. Phys. 1998. V. 84. № 11. P. 6268.
Nakashima S., Harima H. // Physica Status Solidi (a). 1997. V. 162. № 1. P. 39.
Yang B., Cai W., He P. et al. // J. Appl. Phys. 1995. V. 77. № 12. P. 6733.
Chen J., Steckl A.J., Loboda M.J. // Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures Processing, Measurement, and Phenomena. 1998. V. 16. № 3. P. 1305.
Moro L., Paul A., Lorents D.C. et al. // J. Appl. Phys. 1997. V. 81. № 9. P. 6141.
GaussView. Version 6. 2016.
Hisao Nakashima T.S. // Japan. J. Appl. Phys. 1966. V. 5. № 10. P. 874.
Kim K., Park C., Roh J. et al. // J. Vacuum Science & Technology A: Vacuum, Surfaces, and Films. 2001. V. 19. № 5. P. 2636.
Jusoh W.Z.A.W., Rahman S.A., Ahmad A.L., Mokhtar N.M. // Comptes Rendus Chimie. 2019. V. 22. № 11. P. 755.
Wang Y., Fang Z., Zhao S. et al. // RSC Advances. 2018. V. 8. №. P. 22469.
Pacheco F., Sougrat R., Reinhard M. et al. // J. Membrane Science. 2016. V. 501. № 1. P. 33.
Bosi M., Ferrari C., Nilsson D., Ward P. // CrystEngComm. 2016. V. 18. № 39. P. 7478.
Madapura S., Steckl A., Loboda M. // J. Electrochem. Soc. 1999. V. 146. № 3. P. 1197.
Scholz R., Gösele U., Niemann E. et al. // Diamond and Related Materials. 1997. V. 6. № 10. P. 1365.
Fitzer K.M., Schaefer W. The Chemistry of the Pyrolytic Conversion of Organic Compounds to Carbon in Chemistry and Physics of Carbon / Ed. by P.L. Walker, New York: Marcel Dekker Inc., 1971. V. 7. P. 329.
Dufour G., Rochet F., Stedile F.C. et al. // Phys. Rev. B. 1997. V. 56. № 7. P. 4266.
Pezoldt J., Cimalla V. // Crystals. 2020. V. 10. № 6. P. 523.
Kuzmina V., Soldatenko S., Sinelnikov A. // Alternative Energy and Ecology (ISJAEE). 2018. P. 96.
Teker K., Lee K.H., Jacob C. et al. // MRS Proceedings. 2011. V. 640. P. H5.10. https://doi.org/10.15518/isjaee.2018.22-24.096-106
Learn A.J., Khan I.H. // Thin Solid Films. 1970. V. 5. № 3. P. 145.
Иевлев В.М., Кущев С.Б., Солдатенко С.А. и др. // Поверхность. Рентгеновские, cинхротронные и нейтронные исследования. 2009 № 10. С. 48.
Jurkiewicz K., Pawlyta M., Burian A. // J. Carbon Res. 2018. V. 4. №. P. 68.
Bantaculo R., Fukidome H., Suemitsu M. // IOP Conference Series: Materials Science and Engineering. 2015. V. 79. № 1. P. 012004.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии