

Журнал физической химии, 2021, T. 95, № 7, стр. 1059-1067
Конструирование полного набора гомодесмических реакций с использованием метода DEPTH-FIRST SEARCH
С. Л. Хурсан a, *, А. С. Исмагилова b, **, Ф. Т. Зиганшина c, ***, А. И. Ахметьянова b, ****
a Российская академия наук, Уфимский федеральный исследовательский центр,
Уфимский институт химии
Уфа, Россия
b Башкирский государственный университет
Уфа, Россия
c Уфимский государственный нефтяной технический университет
Уфа, Россия
* E-mail: khursansl@anrb.ru
** E-mail: ismagilovaas@yandex.ru
*** E-mail: fairusa85@mail.ru
**** E-mail: ai-albina@mail.ru
Поступила в редакцию 26.09.2020
После доработки 10.11.2020
Принята к публикации 12.11.2020
Аннотация
Предложен теоретико-графовый алгоритм, основанный на процедуре анализа графов Depth-First Search (DFS), для определения полного набора гомодесмических реакций (ГДР) органических соединений. Полный набор ГДР формируется при помощи декомпозиции молекулярного графа исследуемого соединения на ряд более простых составляющих. На основе разработанного DFS-алгоритма реализована программа для построения базиса ГДР, включающая автоматизированную генерацию строения участников гомодесмической реакции и первичный термохимический анализ: расчет тепловых эффектов ГДР из абсолютных энтальпий участников реакции, рассчитанных квантово-химическим методом, определяемым пользователем. Работа алгоритма проиллюстрирована на примере винилциклопропана. Проанализированы достоинства использования полных наборов ГДР для термохимического анализа молекулярной энергетики: высокая точность, возможность контроля воспроизводимости результатов и отсева малооправданных или ошибочных термохимических данных, количественный учет невалентных эффектов строения органических соединений и блочный анализ термохимии близких по строению групп органических соединений, идейно аналогичный известной концепции “активных термохимических таблиц”.
Концепция изодесмичности – сохранения числа и типа химических связей в формально подобранном “превращении” одного набора соединений в другой – была предложена еще в начале 70-х годов прошлого века [1]. Данный подход оказался весьма плодотворным для анализа взаимосвязи между строением органических соединений и их энергосодержанием. Достаточно быстро было выявлено, что качество расчетов можно повысить, дополнив набор балансов изодесмической реакции требованием группового баланса, т.е. одинакового ближайшего валентного окружения каждого атома в формальной реакции. Такой подход был назван гомодесмическим [2]. Вообще, изо- и гомодесмическая концепции с успехом применялись для описания термохимических свойств как отдельных молекул, так и некоторых классов органических соединений [3–7]. Также в ряде работ объектом исследования были и невалентные эффекты, главным образом, энергии напряжения циклов [8, 9]. Качество термохимических оценок улучшалось главным образом благодаря стремительному развитию средств вычислений и появлению более эффективных вычислительных методов квантовой химии. В цитируемых работах выбор изодесмической реакции был произволен и не всегда оптимален, что приводило к существенным расхождениям в результатах термохимических оценок.
Гомодесмический алгоритм предполагает сравнительный термохимический анализ энергетики исследуемых соединений, т.е. для проведения такого анализа необходима априорная информация об энергетических величинах соединений сравнения, полученная либо экспериментально, либо с помощью надежных теоретических вычислений. Ненадежность реперных величин – один из главных источников погрешности гомодесмических оценок, причем зачастую достоверность экспериментальных величин установить невозможно ввиду ограниченности сведений о термохимических свойствах органических соединений.
В настоящей работе использован теоретико-графовый подход для анализа энергетики органических соединений. Ранее близкие идеи использования матриц смежности для подбора изодесмических реакций использовал Фиштик [10, 11], однако в цитируемых работах даже не обсуждается идея применения такого представления для формирования полного набора изо- или гомодесмических процессов. Теоретико-графовый подход авторов [12, 13] учитывает отмеченные выше недостатки. Во-первых, декомпозиция графа исследуемого соединения всеми возможными способами позволит конструировать гомодесмические реакции (ГДР), в которых воспроизводится и дальнее окружение, что является логическим развитием гомодесмической концепции и позволяет изучать валентные и невалентные эффекты строения в рамках одной расчетной схемы. Во-вторых, разнообразие ГДР обеспечивает множественность оценок термохимической величины, что позволяет контролировать воспроизводимость результатов и отсеивать ненадежные расчетные данные. В-третьих, анализ полного набора ГДР для исследуемого соединения формирует некоторый блок соединений, энтальпии образования которых взаимосвязаны через комбинации различных ГДР. Представляется, что такой “блочный” анализ энергетики органических соединений будет более надежным благодаря самосогласованности всех элементов блока. Следует добавить, что близкие идеи совместного анализа термодинамических свойств широкого круга органических соединений развиваются авторами так называемых активных термохимических таблиц [14].
Практическое воплощение концепции полного набора гомодесмических реакций требует разработки соответствующего программного обеспечения для компьютерного анализа строения исследуемого соединения и автоматического конструирования базиса ГДР. В общем случае для произвольного химического соединения существует неоднозначность в выборе ГДР. Определение базиса ГДР позволяет выбирать полную систему независимых термохимических реакций, осуществлять независимые оценки и контролировать воспроизводимость результатов.
В работах [12, 13, 15–18] были представлены этапы конструирования ГДР, их программная реализация на языке программирования Delphi и организация базы данных по химическим соединениям, но не описан модуль получения правых частей ГДР. В разработанной программе для анализа молекулярных графов с целью конструирования гомодесмических реакций использован метод поиска в глубину Depth-First Search (DFS) [19].
Таким образом, основная цель настоящего исследования – развитие теоретико-графового подхода при конструировании гомодесмических реакций путем имплементации метода DFS в алгоритм анализа молекулярных графов и автоматизации подбора референсных соединений в ГДР. Сопутствующая цель работы – апробация усовершенствованного алгоритма на примере винилциклопропана, расчет энергетических характеристик (энергия напряжения цикла, энтальпии образования) исследуемого соединения и референсных структур.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
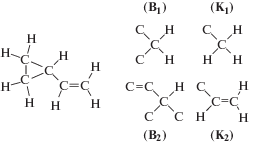
Для иллюстрации работы программного приложения рассмотрим основные этапы процесса конструирования базиса ГДР на примере циклического углеводорода – винилциклопропана C5H8. Применяя аддитивно-групповой метод Бенсона [20], с дополнительным условием неразрывности кратной связи С=С, представим строение винилциклопропана в виде комбинации двух типов внутренних (В1, В2) и одной концевой группы (K2) (рис. 1).
Рис. 1.
Валентная схема винилциклопропана и термохимические группы: внутренние В1, В2 и концевые K1, K2.
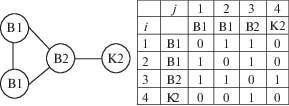
Химические соединения можно представлять в виде графов, вершинами которого являются внутренние и концевые группы, ребрами – химические связи между группами. Для реализации алгоритмов конструирования базиса ГДР на каком-либо языке программирования граф целесообразно представить в виде матрицы смежности вершин. На рис. 2 представлены граф для рассматриваемого соединения и матрица смежности его вершин.
Первый шаг алгоритма конструирования базиса ГДР – формирование всех возможных вариантов матриц смежности вершин, которые соответствуют разрыву связи в графе. Поскольку граф винилциклопропана имеет свойство симметрии (ось симметрии второго порядка), то это означает, что разрывы связей В1–В2 и двойной разрыв (В1–В1)&(В1–В2) могут быть выполнены двумя способами. Разрыв с концевой группой K2 не имеет смысла рассматривать, так как это приводит к вырожденной ГДР. Поэтому в данном алгоритме разрывы с концевыми группами не учитываются. На рис. 3 представлены все возможные варианты разрывов и соответствующие матрицы смежности.
Рис. 3.
Все возможные варианты разрывов химических связей между внутренними группами винилциклопропана и соответствующие матрицы смежности.
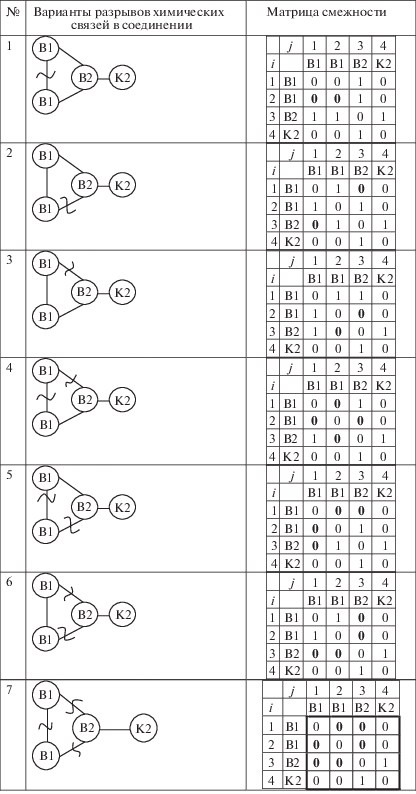
Из рис. 3 видно, что разрывы вида 2 и 3 соответствуют одному типу декомпозиции, аналогичная ситуация имеет место для 4 и 5. Таким образом, мы имеем пять независимых вариантов разрывов и соответственно пять различных вариантов матриц смежности. Каждая полученная матрица смежности соответствует одной ГДР.
На втором шаге, после получения различных вариантов модифицированных матриц смежности выполняется подбор концевых групп. Для слагаемых всех пяти комбинаций подходят концевые группы K1, K2 (рис. 1). Концевая группа конструируется из внутренней группы, центральный атом которой участвует в разрыве химической связи.
Подбор концевых групп осуществляется с помощью расширения исходной матрицы с учетом связности добавленных концевых групп и в соответствии с количеством обнулений в исходной матрице смежности. На рис. 4 представлены графы с концевыми группами и соответствующие расширенные матрицы. Под расширенной матрицей будем понимать матрицу, которая получается путем добавления к исходной матрице строк и столбцов, соответствующих добавленным концевым группам в графе.
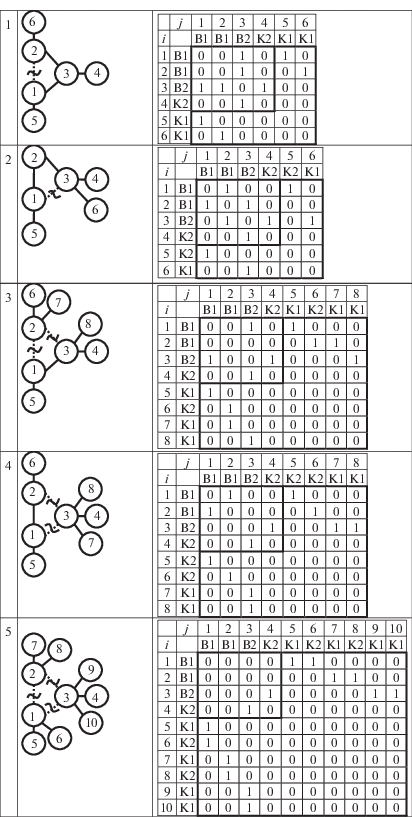
На третьем шаге необходимо сформировать правые части каждой ГДР. Другими словами, необходимо из полученного графа выделить отдельные графы (связные подграфы), из соответствующих расширенных матриц смежности – отдельные подматрицы. Под отдельной подматрицей будем понимать подматрицу, которая соответствует одному компоненту связности в рассматриваемом графе. Каждая выделенная подматрица соответствует продукту ГДР. Разбиение матрицы реализуется с помощью алгоритма на основе метода DFS. Обход графа в глубину DFS позволяет определить вершины, достижимые из определенной вершины. Алгоритм поиска основан на рекурсивном методе, т.е. перебираются все исходящие из рассматриваемой вершины ребра. Если ребро ведет в вершину, которая не была рассмотрена ранее, то запускается алгоритм от этой нерассмотренной вершины, а после возвращаемся и продолжаем перебирать ребра. Возврат происходит в том случае, если в рассматриваемой вершине не осталось ребер, которые ведут в нерассмотренную вершину. Согласно алгоритму таким образом анализируются все вершины исходного графа.
Продемонстрируем работу метода DFS на примере пятой матрицы смежности (рис. 4). Алгоритм начинается со строки, которая соответствует вершине графа. Сначала осуществляется просмотр первой строки – вершины. Из таблицы рис. 4 видно, что первая вершина соединяется с пятой вершиной (в таблице на пересечение первой строки и пятого столбца стоит “1”), а из пятой вершины других исходящих ребер нет, кроме той, которая приводит в первую вершину. Тем самым возвращаемся в первую вершину. Из первой вершины есть еще одно ребро, которое приводит в шестую вершину (в таблице на пересечение первой строки и шестого столбца стоит “1”), а шестая соединяется только с первой вершиной. Из первой вершины других исходящих ребер нет, кроме пятой и шестой. Следовательно, получена компонента связности модифицированного графа с вершинами 1, 5, 6. Аналогично, вторая компонента связности включает вершины 2, 7, 8, третья компонента связности – 3, 4, 9, 10. Таким образом, выделены три компоненты связности, которым соответствуют три независимые подматрицы: B1K1K2; B1K1K2; B2K2K1K1, представленные на рис. 5.
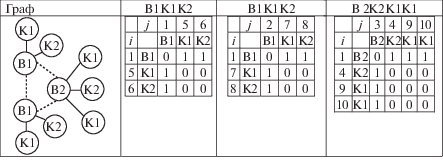
На четвертом этапе на основе полученных подматриц и группового состава формируются правые части ГДР. Номер реакции соответствует типу декомпозиции молекулярного графа, показанного на рис. 4.
(1)
${{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{8}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{1}}}} \to {{{\text{K}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{2}}}}{{{\text{K}}}_{{\text{2}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{1}}}},$(2)
${{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{8}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}} \to {{{\text{K}}}_{{\text{2}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{2}}}}{{{\text{K}}}_{{\text{2}}}}{{{\text{K}}}_{{{\text{11}}}}},$(3)
${{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{8}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{1}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}} \to {{{\text{K}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{2}}}}{{{\text{K}}}_{{\text{2}}}}{{{\text{K}}}_{{\text{1}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}},$(4)
${{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{8}}}} + 2{{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}} \to {{{\text{K}}}_{{\text{2}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{2}}}}{{{\text{K}}}_{{\text{2}}}},$(5)
${{{\text{C}}}_{{\text{5}}}}{{{\text{H}}}_{{\text{8}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{1}}}} + {\text{2}}{{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}} \to 2{{{\text{K}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{2}}}} + {{{\text{K}}}_{{\text{1}}}}{{{\text{K}}}_{{\text{1}}}}{{{\text{В}}}_{{\text{2}}}}{{{\text{K}}}_{{\text{2}}}}.$На пятом этапе после формирования правых частей ГДР происходит инициализация полученных ГДР, т.е. программа определяет соединения и их названия, используя соответствующую базу данных [16]:
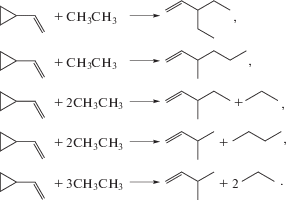
Если соединение отсутствует в базе данных, то она пополняется информацией о названии, химическом и групповом составе соединения, энтальпии образования, а также об абсолютной энтальпии, рассчитанной квантово-химически с помощью нескольких уровней теории, по умолчанию – B3LYP/6-31G(d), M06-2X/aug-cc-pVTZ, G3 и G4. Для всех термохимических уравнений полного набора ГДР рассчитывается тепловой эффект, который служит первичной термохимической информацией для более глубокого анализа и выявления взаимосвязи “строение–свойство”.
В частности, в пяти ГДР для винилциклопропана использованы восемь углеводородов, для пяти из которых в табл. 1 представлена известная из литературы информация о газофазных стандартных энтальпиях образования (ΔfH°) – этана, пропана, бутана, 3-метилбутена-1 и винилциклопропана [21]. Причем для последнего соединения приведены два различающихся значения: 127 [22] и 131.2 ± 1.4 кДж/моль [23]. Ввиду нехватки необходимых данных, проверить достоверность этих значений можно только с использованием ГДР (4) и (5) с помощью тепловых эффектов этих реакций, рассчитанных композитными методами G3 [24] и G4 [25] (табл. 2). Все расчеты проведены с использованием программного пакета Gaussian-09 [26]. Две независимые оценки дают согласующиеся результаты, свидетельствующие о большей надежности экспериментального значения ΔfH° винилциклопропана, полученного авторами [22]. Таким образом, развиваемый нами подход позволяет обоснованно отсеивать ошибочные и недостоверные данные по термохимическим свойствам органических соединений. Более того, информации, приведенной в табл. 1 и 2, достаточно для надежной оценки энтальпии образования трех оставшихся алкенов. Используя найденную нами энтальпию образования винилциклопропана и тепловые эффекты ГДР (1–3), мы рассчитали отсутствующие в [21] величины (табл. 1). Отметим, что два композитных метода дают практически совпадающие результаты.
Таблица 1.
Абсолютные энтальпии соединений (Хартри), рассчитанные композитными методами G3 и G4 и стандартные энтальпии образования (кДж/моль)
| Соединение | $H_{{298}}^{^\circ }$ (G3) | $H_{{298}}^{^\circ }$ (G4) | ${{\Delta }_{{\text{f}}}}H_{{298}}^{^\circ }$ | Примечание |
|---|---|---|---|---|
| Винилциклопропан | –195.096133 | –195.132641 | 128.8 (G3) | Среднее по ГДР (4) и (5) |
| 127.1 (G4) | ||||
| Этан | –79.718910 | –79.733661 | –84.0 | [21] |
| Пропан | –118.988316 | –119.010256 | –104.7 | [21] |
| Бутан | –158.257936 | –158.287099 | –125.6 | [21] |
| 3-Метилбутен-1 | –196.318720 | –196.354936 | –25.5 | [21] |
| 3-Метилпентен-1 | –235.588738 | –235.632153 | –47.6 | ГДР (3) |
| 3-Метилгексен-1 | –274.858479 | –274.909094 | –69.2 | ГДР (2) |
| 3-Этилпентен-1 | –274.858699 | –274.909336 | –69.8 | ГДР (1) |
| 3-Метил-циклопентен-1 | –234.402997 | –234.445859 | 9.67; 7.36; 8.0 | [21] |
| Пропен | –117.777073 | –117.798805 | 20.41 | [21] |
| Бутен-1 | –157.046169 | –157.075059 | –0.63 | [21] |
| Бутен-2 | –157.050305 | –157.079280 | –10.8 | [21] |
| Пентен-2 | –196.319555 | –196.355694 | –32.1 | [21] |
Таблица 2.
Тепловые эффекты гомодесмических реакций (кДж/моль), рассчитанные композитными методами G3 и G4
| ГДР | G3 | G4 |
|---|---|---|
| 1 | –114.6 | –113.0 |
| 2 | –114.0 | –112.4 |
| 3 | –113.2 | –111.4 |
| 4 | –112.1 | –110.5 |
| 5 | –111.6 | –109.8 |
| RSE* | 115.6 ± 0.6 | 114.1 ± 0.6 |
Еще одно достоинство полного набора ГДР – возможность анализа невалентных эффектов строения органических соединений. В структуре винилциклопропана присутствует напряженный трехчленный цикл, поэтому тепловые эффекты ГДР сильно отличаются от нуля и служат надежной оценкой (с обратным знаком) величины энергии напряжения цикла RSE. Из табл. 2 видно, что тепловые эффекты всех ГДР полного набора согласуются друг с другом, что позволяет с высокой степенью достоверности оценить энергию напряжения цикла в винилциклопропане. Следует учесть, что тепловые эффекты ГДР могут быть отягощены минорными эффектами строения (гош-, водородная связь и т.д.), подробно проанализированными в недавних работах [27, 28]. В случае винилциклопропана таким минорным является эффект малых молекул, проявляющийся в линейной зависимости теплового эффекта от числа молекул этана в ГДР. По-видимому, этот эффект обусловлен вычислительными проблемами квантовой химии, предположительно, частичной размерной несогласованностью используемых методов. В практических оценках эффект малых молекул может быть легко учтен с помощью линейного корреляционного анализа, результат представлен в табл. 2.
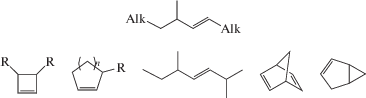
Наконец, следует отметить еще один полезный практический аспект использования полных наборов ГДР в термохимическом анализе. Энтальпии образования восьми органических соединений – участников ГДР (1–5) – образуют посредством энтальпии образования и энергии напряжения цикла винилциклопропана внутренне согласованный блок термохимической информации. Такие же согласованные блоки в форме полных наборов ГДР могут быть получены для других органических соединений, при этом любые два блока могут иметь одинаковые элементы – органические соединения, являющиеся реагентом или продуктом гомодесмических реакций в обоих блоках. Таким образом, совместный термохимический анализ двух и более полных наборов ГДР позволяет еще более повысить надежность термохимических оценок и выстроить термохимическую картину широкого набора органических соединений по блочному типу подобно популярной игре-развлечению PUZZLE. Блочный анализ термохимии органических соединений во многом аналогичен развиваемой концепции активных термохимических таблиц [15]. Рассмотрим в качестве иллюстрации один из продуктов ГДР (3) – 3-метилпентен-1. Это соединение – продукт декомпозиции винилциклопропана по методике, изложенной выше, – содержит в своем составе комбинацию внутренних групп В1В2 (рис. 1). Соответственно полные наборы ГДР для широкого круга органических соединений будут включать в себя 3-метилпентен-1, если эти соединения содержат фрагмент, приведенный на рис. 6. Там же представлены примеры таких соединений.
Рис. 6.
Субструктура, содержащая внутренние группы В1В2, и примеры соединений, при декомпозиции которых алгоритм DFS будет генерировать данную субструктуру.
Например, в работе [27] приведен полный набор ГДР для 3‑метил-циклопентена‑1, две реакции из этого набора включают 3‑метилпентен‑1 в качестве реперного соединения:
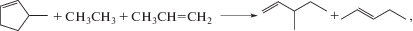
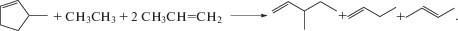
Стандартные энтальпийные величины указанных в (6) и (7) соединений приведены в табл. 1. Для энтальпии образования 3‑метил-циклопентена‑1 приведены три значения из базы данных [21], для дальнейших расчетов использована средняя величина 8.3 кДж/моль. С использованием данных композитных расчетов методами G3 и G4 и приведенных уравнений мы получаем четыре независимых оценки энтальпии образования 3‑метилпентена‑1 (–47.8 ± 0.3 кДж/моль), которые прекрасно согласуются друг с другом и с расчетами для соединений винилциклопропанового блока (табл. 1). Очевидно, что совместный термохимический анализ системы взаимопересекающихся блоков – наборов органических соединений – позволяет выстроить внутренне согласованную термохимическую картину для широкого круга органических соединений самых различных классов.
В результате выполненной работы усовершенствована методика теоретико-графового подхода к конструированию гомодесмотических реакций благодаря использованию Depth-First Search метода анализа матриц смежности. Применение алгоритма DFS к гомодесмическому анализу органических соединений дает возможность простого, алгоритмизированного построения полных наборов гомодесмических реакций. Новая методика проиллюстрирована на примере формирования полного набора ГДР винилциклопропана, для которого определена энергия напряжения цикла (114.1 ± 0.6 кДж/моль, G4), а также рассчитаны энтальпии образования винилциклопропана и ряда референсных соединений, образующие внутренне (в изученном наборе) и внешне (с надежными литературными данными) согласованный массив термохимической информации. В совокупности с квантово-химическими оценками энтальпийных характеристик разработанный алгоритм служит универсальным теоретическим методом расчета и прогнозирования термохимических свойств химических соединений, который дает возможность надежного определения тепловых эффектов химических реакций, энтальпий образования, энергетических эффектов строения молекул, что, помимо фундаментальных представлений, может быть использовано при разработке технологических регламентов и осознанного управления практически важными химическими процессами.
Все квантово-химические вычисления проводили на оборудовании центра коллективного пользования “Химия” Уфимского Института химии УФИЦ РАН и РЦКП “Агидель” УФИЦ РАН. Работа выполнена при финансовой поддержке РФФИ, грант № 18-07-00584 А, а также в рамках государственного задания по теме НИР УфИХ УФИЦ РАН АААА-А20-120012090030-6.
Список литературы
Hehre W.J., Ditchfield R., Radom L., Pople J.A. // J. Amer. Chem. Soc. 1970. V. 92. № 16. P. 4796.
A Hierarchy of Homodesmotic Reactions for Thermochemistry (S.E. Wheeler, K.N. Houk, P. v. R. Schleyer, W.D. Allen) Modern Electronic Structure Theory, Part 1 // D. Varkony // World Scientific. 1995. P. 1462. https://doi.org/10.1021/ja805843n
Хурсан С.Л. // Журн. физ. химии. 2002. Т. 76. № 3. С. 481. Khursan S.L. // Russ. J. Phys. Chem. A. 2002. V. 76. № 3. P. 405.
Khursan S.L. // Russ. J. Phys. Chem. A. 2004. V. 78. Suppl. 1. P. S34.
Шайхлисламов Д.С., Талипов М.Р., Хурсан С.Л. // Журн. физ. химии. 2007. V. 81. №. 2. P. 293. Shaikhlislamov D.S., Talipov M.R., Khursan S.L. //Russ. J. Phys. Chem. A. 2007. V. 81. № 2. P. 235. https://doi.org/10.1134/S003602440702015X
Dorofeeva O.V., Ryzhova O.N., Moiseeva N.F. // Russ. J. Phys. Chem. A. 2008. V. 82. № 6. P. 933. https://doi.org/10.1134/S0036024408060113
Dorofeeva O.V., Ryzhova O.N., Suntsova M.A. // J. Phys. Chem. A. 2013. V. 117. № 31. P. 6835. https://doi.org/10.1021/jp404484q
Bachrach S.M. // J. Chem. Educ. 1990. V. 67. № 11. P. 907. https://doi.org/10.1021/ed067p907
Dudev T., Lim C. // J. Am. Chem. Soc. 1998. V. 120. № 18. P. 4450.
Fishtik I. // J. Phys. Chem. A. 2012. V. 116. № 7. P. 1854. https://doi.org/10.1021/jp211795s
Vijaya P., Tadéa M.O., Fishtik I., Dattab R. // Computers and Chemical Engineering. 2013. № 49. P. 85.
Хурсан С.Л., Исмагилова А.С., Ахмеров А.А., Спивак С.И. // Журн. физ. химии. 2016. Т. 90. № 4. С. 569. https://doi.org/ Khursan S.L., Ismagilova A.S., Akhmerov A.A., Spivak S.I. // Russ. J. Phys. Chem. A. 2016. V. 90. № 4. P. 796. https://doi.org/10.1134/S003602441603020110.1134/S0036024416030201https://doi.org/10.7868/S0044453716030201
Хурсан С.Л., Исмагилова А.С., Спивак С.И. // Докл. АН. 2017. Т. 474. № 4. С. 454. https://doi.org/ Khursan S.L., Ismagilova A.S., Spivak S.I. // Doklady Physical Chemistry. 2017. V. 474. № 2. P 99. https://doi.org/10.1134/S001250161706003310.1134/S0012501617060033https://doi.org/10.7868/S0869565217040120
Ruscic B., Pinzon R.E., Morton M.L. et al. // J. Phys. Chem. A. 2004. V. 108. P. 9979.
Зиганшина Ф.Т., Ахметьянова А.И., Исмагилова А.С. и др. // Системы управления и информационные технологии. 2018. № 4 (74). С. 72.
Зиганшина Ф.Т., Исмагилова А.С., Ахметьянова А.И. и др. // Там же. 2019. № 4 (78). С. 10.
Ахмеров А.А., Исмагилова А.С., Спивак С.И., Хурсан С.Л. Конструирование гомодесмических реакций и расчет энтальпии образования органических соединений: Свидетельство о гос. регистрации программы для ЭВМ № 2015617060. 29.06.2015.
Ахмеров А.А., Исмагилова А.С., Спивак С.И., Хурсан С.Л. Энергетические характеристики органических соединений: Свидетельство о гос. регистрации базы данных № 2015621003. 01.07.2015.
Кормен Т.Х., Лейзерсон Ч.И., Ривест Р.Л., Штайн К. Алгоритмы: построение и анализ. 3-е изд. Издательский дом “Вильямс”, 2013. 1328 с. ISBN 978-5-8459-1794-2 (рус.). ISBN 978-0-2620-3384-8 (англ.). Cormen T.H., Leiserson C.E., Rivest R.L., Stein C. Introduction to Algorithms. Third Edition. The MIT Press. 2009. 1313 p. ISBN 978-0-2620-3384-8
Бенсон С.У. Термохимическая кинетика. М.: Мир, 1971. 308 с. Benson S.W. Thermochemical Kinetics. New York: Wiley, 1968.
Afeefy H.Y., Liebman J.F., Stein S.E. “Neutral Thermochemical Data” in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, Ed. by P.J. Linstrom and W.G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, https://doi.org/10.18434/T4D303, (retrieved September 1, 2020).
Roth W.R., Adamczak O., Breuckmann R. et al. // Chem. Ber. 1991. V. 124. P. 2499.
Лебедева Н.Д., Гутнер Н.М., Назарова Л.Ф. // Термодинамика органических соединений, Межвузовский сборник, ГГУ им. Н.И. Лобачевского / Под ред. И.Б. Рабиновича. 1977. Вып. 6. С. 26.
Curtiss L.A., Raghavachari K., Redfern P.C. et al. // J. Chem. Phys. 1998. V. 109. P. 7764.
Curtiss L.A., Redfern P.C., Raghavachari K. // Ibid. 2007. V. 126. P. 84.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., K.N. Kudin, Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J., Gaussian 09, Revision C.1, Gaussian Inc., Wallingford CT, 2009.
Akhmetshina E.S., Khursan S.L. // Thermochim. Acta. 2020. V. 685. P. 178541. https://doi.org/10.1016/j.tca.2020.178541
Ахметшина Е.С., Хурсан С.Л. // Изв. АН. Сер. хим. 2020. № 1. С. 76. Akhmetshina E.S., Khursan S.L. // Russ. Chem. Bulletin. 2020. V. 69. № 1. Р. 76. https://doi.org/10.1007/s11172-020-2725-7
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии