

Журнал физической химии, 2021, T. 95, № 9, стр. 1370-1374
Алгоритм расчета констант протолитических равновесий органических кислот в изопропиловом спирте
С. С. Лысова a, b, *, Т. А. Скрипникова a, b, Ю. Э. Зевацкий a, b, c
a Санкт-Петербургский государственный университет промышленных технологий и дизайна
191186 Санкт-Петербург, Россия
b АО “Новбытхим”
188300 г. Гатчина, Ленинградская область, Россия
c Санкт-Петербургский государственный технологический институт (технический университет)
190013 Санкт-Петербург, Россия
* E-mail: lusovass@mail.ru
Поступила в редакцию 09.12.2020
После доработки 09.12.2020
Принята к публикации 11.01.2021
Аннотация
Разработан алгоритм расчета констант диссоциации слабых органических кислот в изопропиловом спирте на основе спектрофотометрических данных в УФ- и видимой областях без измерений рН среды и без использования буферных растворов. Предложен новый способ определения аналитических длин волн в спектрофотометрии. Предлагаемый алгоритм апробирован экспериментально для семи органических кислот различной силы; относительная погрешность определения констант диссоциации не превышает 1%.
Ранее нами были предложены новые алгоритмы расчета термодинамических констант диссоциации для слабых органических кислот, оснований [1], амфолитов [2] и более сложных объектов [3, 4] в водных растворах с использованием концентрационной УФ/вид-спектрофотометрии. По сравнению со стандартными методиками спектрофотометрического и потенциометрического pH-титрований, этот метод не требует измерений значений pH среды и использования буферных растворов с постоянной ионной силой, что является существенной проблемой в неводных средах.
В настоящей работе мы расширили возможности данного подхода и применили его к неводным средам для определения точных значений термодинамических констант диссоциации pKа органических кислот различной силы. В качестве неводной среды был выбран изопропиловый спирт – один из наиболее используемых в титрованиях растворитель [5]. Благодаря своей дешевизне и низкой полярности он позволяет эффективно растворять большинство органических соединений. Кроме того, изопропиловый спирт имеет подходящие спектральные характеристики.
Известно, что чувствительность и погрешность спектрофотометрического определения зависят от аналитической длины волны, при которой проводятся измерения. Традиционно аналитическую длину волны выбирают таким образом, чтобы она соответствовала наибольшей разности между оптическими плотностями растворов крайних прототропных форм [6, 7]. В настоящей работе мы предложили новый способ определения оптимальной длины волны без использования концентрированных растворов сильных кислот и оснований.
ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
В качестве первичных экспериментальных данных использовали зависимости D (оптическая плотность) – С (концентрация исследуемого вещества), которые получали с помощью УФ-спектров исследуемой органической кислоты при различных концентрациях в изопропиловом спирте с добавлением гидроксида натрия. Оптимальную концентрацию гидроксида натрия определяли по методике, описанной в [1]. Концентрация гидроксида натрия – постоянная величина и, как правило, на порядок меньше концентрации исследуемой кислоты (не превышает значений 10–3 моль/л). В таких растворах с очень низкой ионной силой (I → 0) можно пренебречь межмолекулярными взаимодействиями и ассоциацией, считая их близкими к идеальным, что приводит к совпадению значений концентрационных констант с термодинамическими.
Общая схема процессов диссоциации слабой кислоты АН в присутствии гидроксида натрия и образующейся соли NaА, а также воды, содержащейся в изопропиловом спирте, описывается равновесиями:
Согласно закону действующих масс, константы равновесий этих процессов выражаются через равновесные концентрации соответствующих частиц:
(1)
${{K}_{{\text{a}}}} = \frac{{[{{{\text{A}}}^{ - }}][{{{\text{H}}}^{ + }}]}}{{\left[ {{\text{AH}}} \right]}},$(2)
${{K}_{{\text{b}}}} = \frac{{[{\text{N}}{{{\text{a}}}^{{\text{ + }}}}][{\text{O}}{{{\text{H}}}^{ - }}]}}{{\left[ {{\text{NaOH}}} \right]}},$(3)
${{K}_{{\text{S}}}} = \frac{{[{\text{N}}{{{\text{a}}}^{{\text{ + }}}}][{{{\text{A}}}^{ - }}]}}{{\left[ {{\text{NaA}}} \right]}},$Решая совместно уравнения материального баланса:
(6)
${{С}_{{\text{b}}}} = [{\text{N}}{{{\text{a}}}^{{\text{ + }}}}] + [{\text{NaOH}}] + [{\text{NaA}}],$(7)
$[{{{\text{Н}}}^{{\text{ + }}}}] + [{\text{N}}{{{\text{a}}}^{ + }}] = [{\text{O}}{{{\text{H}}}^{ - }}] + [{{{\text{A}}}^{ - }}],$(8)
$[{{{\text{Н}}}^{{\text{ + }}}}] + [{\text{N}}{{{\text{a}}}^{{\text{ + }}}}] - \frac{{{{{\text{K}}}_{{\text{i}}}}}}{{[{{{\text{Н}}}^{{\text{ + }}}}]}} - \frac{{{{С}_{{\text{i}}}}}}{{1 + \frac{{[{\text{N}}{{{\text{a}}}^{{\text{ + }}}}]}}{{{{K}_{{\text{S}}}}}} + \frac{{[{{{\text{Н}}}^{{\text{ + }}}}]}}{{{{K}_{{\text{a}}}}}}}} = 0,$(9)
$[{{{\text{A}}}^{ - }}] = \frac{{{{С}_{{\text{i}}}}}}{{1 + \frac{{[{\text{N}}{{{\text{a}}}^{{\text{ + }}}}]}}{{{{K}_{{\text{S}}}}}} + \frac{{[{{{\text{Н}}}^{{\text{ + }}}}]}}{{{{K}_{{\text{a}}}}}}}},$(10)
$\left[ {{\text{AH}}} \right] = \frac{{[{{{\text{A}}}^{ - }}][{{{\text{H}}}^{ + }}]}}{{{{K}_{{\text{a}}}}}},$(11)
$\left[ {{\text{NaA}}} \right] = \frac{{[{\text{N}}{{{\text{a}}}^{{\text{ + }}}}][{{{\text{A}}}^{ - }}]}}{{{{K}_{{\text{S}}}}}}.$Оптимальную аналитическую длину волны λопт определяли следующим образом. В рабочем диапазоне оптической плотности (D от 0.2 до 2) снимали спектры исследуемой кислоты при двух разных концентрациях Сmin (D = 0.2) и Сmax (D = = 2). Рассчитывали молярные коэффициенты поглощения при Сmin и Сmax на всех длинах волн. Оптимальной аналитической длиной волны λопт будет та, при которой максимально различаются молярные коэффициенты поглощения при Сmin и Сmax.
Если имеется несколько равноценных, с этой точки зрения длин волн, предпочтение следует отдать той из них, которая расположена в длинноволновой области. Все дальнейшие измерения проводили при одной оптимальной длине волны.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объекты исследования
Пикриновую кислоту, 4-нитробензойную кислоту, 3,5-динитробензойную кислоту, 3-нитрофенол и 4-нитрофенол квалификации “ч.д.а.” использовали без дополнительной очистки. Бензойную кислоту и фенол квалификации “х.ч.” очищали возгонкой при атмосферном давлении. Чистоту веществ контролировали по температуре плавления. Изопропиловый спирт квалификации “ос.ч.” использовали без дополнительной очистки. Чистоту контролировали по УФ-спектрам поглощения.
Концентрационная УФ/вид-спектрофотометрия
Серию рабочих растворов с различной концентрацией исследуемой кислоты готовили весовым методом разбавления. Навеску исходного вещества и его растворы взвешивали на аналитических весах MBA210-A с точностью ±0.1 мг. Спектры поглощения регистрировали на спектрофотометре Shimadzu 2700, используя кварцевые кюветы размером 0.1 и 1 см. Контроль температуры исследуемого раствора и кюветы сравнения осуществляли с помощью держателя кювет Shimadzu ТСС-100 с термоэлектрическим контролем температуры с точностью ±0.1 K. Все измерения проводили при температуре 25°C.
Значения плотности рабочих растворов исследуемых кислот измеряли с помощью ультразвукового плотномера DMA-5000 c абсолютной погрешностью измерений ±5 × 10–6 г/см3. Встроенный термостат позволял проводить определения плотности растворов при 25°C. Точность поддержания температуры составляла ±0.01 K.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Предлагаемый алгоритм апробирован экспериментально для пикриновой кислоты, бензойной кислоты, 3,5-динитробензойной кислоты, 4-нитробензойной кислоты, фенола, 3-нитрофенола и 4-нитрофенола. Мы анализировали спектры поглощения исследуемых веществ при различных концентрациях, используя концентрационную УФ/вид-спектрофотомерию.
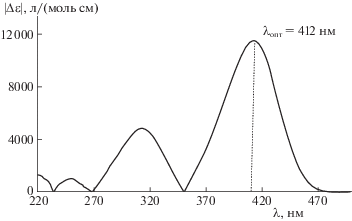
На рис. 1 представлены спектры поглощения 4-нитрофенола при различных концентрациях в присутствии гидроксида натрия с постоянной концентрацией. Оптимальную длину волны выбирали по максимальной разности молярных коэффициентов поглощения 4-нитрофенола при двух различных концентрациях, как показано на рис. 2. Обрабатывая по предлагаемому алгоритму первичные данные Di (оптическая плотность) – Сi (концентрация 4-нитрофенола) при одной оптимальной длине волны λопт, определяли неизвестные значения pKa, pKi, pKS, ${{\varepsilon }_{{{{{\text{A}}}^{ - }}}}}$, εHA, εNaA и Cb.
Рис. 1.
Спектры поглощения 4-нитрофенола при различных концентрациях с добавлением NaOH (Сb = 2.0 × 10–4 моль/л) в изопропиловом спирте; Ci (моль/л): 1 – 2.0 × 10–3, 2 – 1.9 × 10–3, 3 – 1.8 × 10–3, 4 – 1.7 × 10–3, 5 – 1.6 × 10–3, 6 – 1.5–10–3, 7 – 1.4 × 10–3, 8 – 1.3 × 10–3, 9 – 1.2 × 10–3, 10 – 1.1 × 10–3, 11 – 1.0 × 10–3, 12 – 9.5 × 10–4, 13 – 8.5 × 10–4, 14 – 7.7 × × 10–4, 15 – 6.9 × 10–4, 16 – 5.9 × 10–4, 17 – 5.3 × 10–4, 18 – 4.6 × 10–4, 19 – 4.2 × 10–4, 20 – 3.6 × 10–4, 21 – 3.3 × 10–4.

Рис. 2.
Выбор оптимальной длины волны λопт по разности молярных коэффициентов поглощения 4-нитрофенола при концентрациях Cmax = 2.0 × 10–3 моль/л и Cmin = 3.3 × 10–4 моль/л с добавлением NaOH (Сb = 2.0 × 10–4 моль/л) в изопропиловом спирте.
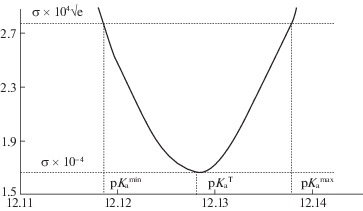
Погрешность определения термодинамической константы диссоциации ${\text{p}}K_{{\text{a}}}^{{\text{T}}}$ для 4-нитрофенола определяли по нормальному закону распределения, как показано на рис. 3, где σ – средняя квадратическая относительная погрешность:
Рис. 3.
Определение погрешности термодинамической константы диссоциации для 4-нитрофенола в изопропиловом спирте (${\text{p}}K_{{\text{a}}}^{{\text{T}}}$ = 12.13 ± 0.01).
В табл. 1 представлены значения ${\text{p}}K_{{\text{a}}}^{{\text{T}}}$ кислот в изопропиловом спирте в сравнении с литературными данными pKa (лит.), а также длина волны λ, количество обработанных спектров N, диапазон концентраций исследуемой кислоты Ci, концентрация гидроксида натрия СNaOH, константа автопротолиза воды в изопропиловом спирте pKi, константа диссоциации соли рKS и значения молярных коэффициентов поглощения нейтральной частицы εAH, анионной ${{\varepsilon }_{{{{{\text{A}}}^{ - }}}}}$ и соли εNaА. Относительная погрешность определения констант диссоциации для всех кислот по предлагаемому алгоритму не превышает 1%.
Таблица 1.
Термодинамические константы диссоциации кислот ${\text{p}}K_{{\text{a}}}^{{\text{T}}}$ в изопропиловом спирте, полученные методом концентрационной УФ/вид спектрофотометрии в сравнении с литературными данными (εi, л/(моль cм))
| Кислота | pKa (лит.) | ${\text{p}}K_{{\text{a}}}^{{\text{T}}}$ | λ, нм | N | Ci, моль/л |
Cb, моль/л |
pKi | pKS | εAH | ${{\varepsilon }_{{{{{\text{A}}}^{ - }}}}}$ | εNaA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Пикриновая кислота | 3.74 [10] 4.02 [11] 4.08 [12] |
3.30 ± 0.01 | 362 | 15 | 1.5 × 10–3–4.3 × 10–4 | 4.6 × 10–4 | 15.01 | 1.67 | 354 | 18 309 | 17 526 |
| Бензойная кислота | 10.2 [13] 11.75 [14] |
10.53 ± 0.05 | 280 | 16 | 3.2 × 10–3–2.3 × 10–4 | 5.6 × 10–4 | 15.12 | 6.92 | 705 | 121 | 124 |
| 3,5-Динитробензойная кислота | 8.31 [10] | 9.03 ± 0.05 | 250 | 23 | 1.2 × 10–3–2.1 × 10–4 | 2.3 × 10–4 | 10.26 | 7.25 | 10 696 | 16 884 | 19 756 |
| 4-Нитробензойная кислота | 9.6 [10] | 10.62 ± 0.04 | 285 | 30 | 1.6 × 10–3–1.2 × 10–4 | 3.7 × 10–5 | 13.82 | 6.66 | 3877 | 30 429 | 17 876 |
| Фенол | 15.41 calc [15] | 15.83 ± 0.07 | 241 | 20 | 5.5 × 10–3–1.6 × 10–4 | 8.2 × 10–4 | 15.91 | 7.81 | 100 | 24 987 | 10 294 |
| 4-Нитрофенол | 11.19 [10] 12.45 [11] |
12.13 ± 0.01 | 412 | 21 | 2.0 × 10–3–3.3 × 10–4 | 2.0 × 10–4 | 13.27 | 7.87 | 78 | 19 071 | 3427 |
| 3-Нитрофенол | 12.65 [10] 13.92 [14] |
11.81 ± 0.08 | 298 | 12 | 1.4 × 10–3–2.3 × 10–4 | 2.7 × 10–4 | 15.56 | 7.19 | 1561 | 13 608 | 2634 |
Следует отметить, что концентрационная УФ/вид-спектрофотометрия позволяет получить значения крайних прототропных форм без использования концентрированных растворов сильных кислот и сильных оснований. Это расширяет возможности спектрофотометрического метода и может быть полезно, когда полный перевод вещества в одну из прототропных форм в неводном растворителе невозможен. Новые исследования показали, что возможности использования концентрационной УФ/вид-спектрофотомерии еще далеко не исчерпаны и требуют дальнейшего изучения.
Список литературы
Lysova S.S., Skripnikova T.A., Zevatskii Yu.E. // Russ. J. Phys. Chem. A. 2017. V. 91. № 12. P. 2366. https://doi.org/10.1134/S0036024417110139
Lysova S.S., Skripnikova T.A., Zevatskii Yu.E. // Ibid. 2018. V. 92. № 5. P. 922. https://doi.org/10.1134/S0036024418050229
Skripnikova T.A., Lysova S.S., Zevatskii Yu.E. // J. Chem. Eng. Data. 2017. V. 62. № 8. P. 2400. https://doi.org/10.1021/acs.jced.7b00308
Skripnikova T.A., Lysova S.S., Zevatskii Yu.E. et al. // J. Mol. Struct. 2018. V. 1154. № 15. P. 59. https://doi.org/10.1016/j.molstruc.2017.10.004
Barbosa J., Bosch E., Suarez F. // Analyst. 1985. V. 110. P. 1473. https://doi.org/10.1039/AN9851001473
Берштейн И.Я., Каминский Ю.Л. Спектрофотометрический анализ в органической химии. Л.: Химия, 1986. 200 с.
Frans S.D., Harris J.M. // Anal. Chem. 1985. V. 57. № 13. P. 2680. https://doi.org/10.1021/ac00290a055
Selitrenikov A.V., Zevatskii Yu.E. // Russ. J. Gen. Chem. 2015. V. 85. № 1. P. 7. https://doi.org/10.1134/S1070363215010028
http://old.exponenta.ru/soft/Mathcad/Mathcad.asp
Chantooni M.K., Kolthoff I.M. // Anal. Chem. 1979. V. 51. № 1. P. 133. https://doi.org/10.1021/ac50037a039
Barbosa J., Bosch C.M., Sanz-Nebot V. // Microchimica Acta. 1992. V. 106. P. 327. https://doi.org/10.1007/BF01242105
Rosés M., Rived F., Bosch E. // J. Chem. Soc., Faraday Trans. 1993. V. 89. P. 1723. https://doi.org/10.1039/FT9938901723
Mollin J., Pavelek Z., Navratilova J., Recmanova A. // Collect. Czech. Chem. Commun. 1985. V. 50. P. 2670. https://doi.org/10.1135/cccc19852670
Bosch E., Rafols C., Roses M. // Anal. Chimica Acta. 1995. V. 302. № 1. P. 109. https://doi.org/10.1016/0003-2670(94)00435-O
Penhoat M. // Tetrahedron Letters. 2013. V. 54. P. 2571. https://doi.org/10.1016/j.tetlet.2013.02.110
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии