

Журнал физической химии, 2021, T. 95, № 9, стр. 1345-1352
Коррекция механизмов перегруппировки Валлаха и побочных реакций
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 23.12.2020
После доработки 23.12.2020
Принята к публикации 03.01.2021
Аннотация
Установлены интермедиаты превращения азоксибензола (AzOB) в изомеры гидроксиазобензола в концентрированной серной кислоте (перегуппировка Валлаха). Учтено, что заряженный азот азогруппы N+ → О– индуцирует в AzOB электронную поляризацию с образованием катиона фениламинильного типа, после присоединения протона к кислороду с образованием группы N+ → ОH следуют акты протонирования второго азота азогруппы с вытеснением гидроксигруппы в виде катиона HO+ и появлением протонированного азобензола AB+H. Установлено, что AB+H вступает в обратимую реакцию “депротонирование ↔ протонирование“ (AB+H ↔ AB + H+) с участием то одного, то другого азота азогруппы; в свою очередь, катионы HO+ быстро взаимодействуют с серной кислотой, создавая гидроксилсульфат-катионы HOSO2O+, реагирующие с нейтральными фенильными кольцами катионов AB+H, а также с серной кислотой. Показано, что конечные продукты перегруппировки – гидроксизамещенные производные AB – вместе с побочными продуктами, включая азобензол, образуются в результате нейтрализации реакционной системы. Дано объяснение кинетики перегруппировки Валлаха, а также путей образования побочных продуктов.
Открытая О. Валлахом в 1880 г. изомеризация азоксибензола (AzOB), катализируемая концентрированной серной кислотой, получила название “перегруппировка Валлаха“ (ПВ) [1, 2]. Она сочетает в себе потерю связанного с азогруппой кислорода и появление гидроксигруппы в пара-позиции бензольного кольца (4-), отдаленного от NO-группы [1, 2]:

Для осуществления ПВ обычно используют серную кислоту в концентрации более 80% при нагревании до T ≥ 50°С. В связи с незначительным выходом (0.02) орто-изомеров (группа OH в позициях 2, 2', 6, 6' на схеме 1), их образование в H2SO4 относят к побочным процессам, причем при наличии заместителей в AzOB и в зависимости от их природы гидроксигруппа может появляться в замещенном или в незамещенном кольце [1, 2].
Согласно обзорной статье [2], реакция ПВ изучена в экспериментальном отношении особенно подробно на примере незамещенного AzOB. Вместе с тем, обзор [2] работ с различными производными азоксибензолов показал наличие побочных процессов к собственно ПВ, и что механизм ПВ более сложен, чем представлялось ранее. При этом можно отметить явную несогласованность в описаниях механизма ПВ в энциклопедии [1] и обзоре [2]. Так, в приведенной в [1] схеме:
протонирование кислорода группы N → O приводит к отщеплению гидрокси-аниона, атакующего углерод в пара-положении фенильного кольца промежуточного дикатиона с вытеснением протона. Однако атака анионом HO– дикатиона PhN+≡N+Ph нереальна в силу высокой скорости нейтрализации HO– при весьма высокой кислотности среды (авторы [3] полагают, что катализатором реакции в таких жестких условиях служат недиссоциированная кислота H2SO4 или ее протонированная форма ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$). К тому же в обзоре [2], опубликованном на год ранее энциклопедии [1], указанная схема не рассматривается, но утверждается участие в ПВ катионоидного интермедиата с неустановленным строением.
Применение AzOB, меченного изотопом 15N по несвязанному с кислородом атому N азогруппы [2, ссылка 9], позволило установить, что оба фенильных кольца гидроксилируются в одинаковой степени:
В связи с этим было предположено образование оксониевого катиона которому в более поздней работе ([2], ссылка 60) приписана роль ключевого участника обмена атома О между азотами азогруппы: Между тем, в [2] приведены возражения против данной схемы с участием в ПВ катиона оксония при азогруппе и участия одних только однопротонированных интермедиатов, но сообщается, что при концентрации [H2SO4] от 75.3 до 99.99% в ПВ должны участвовать дикатионоидные интермедиаты с вероятным строением PhN+≡N+Ph. Кроме того, образование указанного в приведенной выше схеме промежуточного продукта присоединения молекулы воды к хиноидному катиону маловероятно вследствие весьма сильной связи воды с H2SO4 [3, 4].


Относительно участия в ПВ дикатиона PhN+≡N+Ph в [2] предположена следующая картина. Сначала образуется протонированная по кислороду HO-азогруппа. Она тоже протонируется за счет переноса протона от недиссоциированной H2SO4, после чего происходят отщепление воды и образование дикатиона PhN+≡N+Ph, который подвергается быстрой атаке анионом ${\text{HSO}}_{4}^{ - }$ с образованием азобензол-4-бисульфата. Гидролиз последнего дает 4-гидроксиазобензол.
Вместе с тем, в [2] неоднократно подчеркивается, что, несмотря на значительное число работ, посвященных изучению механизма ПВ, единого представления о нем до сих пор не выработано. Остается нерешенным один из ключевых вопросов, заключающийся в идентификации катионоидных частиц, ответственных за протекание перегруппировки. Теперь, благодаря работам [5–13] понятно, что важность накопленных экспериментальных результатов нивелируется устаревшей трактовкой электронного (e-) строения AzOB. В настоящем сообщении дана новая формула e-строения AzOB и показана ее эффективность при объяснении механизма ПВ.
Формула e-строения AzOB
AzOB содержит координационную связь N → О, которая обычно образуется в ситуации, когда молекула одного из реагентов имеет атом с неподеленной e-парой в валентной оболочке (основание Льюиса), а молекула другого реагента (кислота Льюиса) имеет атом с так называемым электронным “пробелом“, например, секстетом электронов вместо устойчивого октета [14, с. 63]. Типичным примером этому служит связь в продукте реакции аммиака с триметилбором H3N: + B(CH3)3, образующаяся за счет того, что принадлежащая азоту e-пара, заполняет “e-пробел“ в атоме бора при фиксации на азоте положительного, а на атоме бора отрицательного заряда.
Другой пример – оксиды аминов, для которых сначала использовали формулу R3N=O, но позже стали записывать в ковалентно-ионном виде с указанием смещения e-пары от азота к кислороду с помощью стрелки R3N → O или в поляризованном виде R3N+–O– [14, с. 65]. Так, для обозначения координационной связи во всех соединениях азоксибензола в обзорной работе [2] используется запись N → O, как в схеме 1. Между тем, исходная квантовая e-конфигурация валентной оболочки (L) атома O (2s22p$_{x}^{2}$2py2pz) [15] изначально не имеет e-пробела, необходимого для образования координационной связи по типу реакции H3N: + B(CH3)3. Поэтому для координационной связи в оксидах аминов R3N → O требуется изменение валентного состояния атома O на промежуточной стадии реакции за счет перехода электрона с одной p-орбитали на другую, например, 2s22p$_{x}^{2}$2py2pz → 2s22p$_{x}^{2}$2pz22py0. В этом случае на кислороде появляется e-пробел (орбиталь $2p_{y}^{0}$), а требуемая для этого некоторая энергетическая затрата должна компенсироваться за счет образования связи R3N → O и трех С–N-связей. Очевидно, что сам факт существования оксидов аминов свидетельствует о наличии такой компенсации.
У молекулы AzOB для стабилизации связи N → O существует дополнительный путь за счет делокализаци заряда по ароматической системе. Он был ранее установлен у нейтральных и протонированных аминоазобензольных красителей [4–8], и с его учетом проведена коррекция механизмов фотохимических реакций для ряда соответствующих соединений [9–13]. Характер такой делокализации заряда показан на примере катиона t-AB+H, образующегося в серной кислоте [4, 11] cхема 2.

Согласно схеме 2, присоединенный к транс-азобензолу (t-AB) протон индуцирует делокализацию вносимого с собой заряда с сохранением sp2-гибридизации атомов азота и плоского строения t-AB+H. При образовании связи H–N+ сначала один из sp2-электронов азота переходит на 1s-орбиталь возникающего атома H, затем оставшийся sp2-электрон азота образует ковалентную связь с 1s-орбиталью водорода, наделяя sp2-орбиталь азота положительным зарядом (показан под азотом на схеме 2). Появившийся на sp2-орбитали протонированного азота заряд притягивает оба π-электрона азогруппы так, что они проводят больше времени на pz-орбитали группы ${\text{HN}}_{ + }^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$. При этом второй атом азота азогруппы приобретает положительный заряд на своей pz-орбитали, превращаясь в катион N+, и если в группе ${\text{HN}}_{ + }^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ противоположные заряды в значительной степени компенсируют друг друга, то положительный заряд возникшего π-катиона N+ частично делокализуется в сопряженном кольце на орто- и пара-положениях, как в катионах бензильного и фениламинильного типа (PhAT, Ph+N+) [4].
Отмеченное явление e-поляризации определяет оптические, кислотно-основные, фотохимические свойства AB+H и большого массива аминоазобензольных красителей [5–13]. Естественно ожидать, что сходное явление характерно и для AzOB и что с ним связаны особенности ПВ.
e-Формула строения AzOB, представленная на схеме 3.
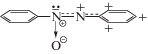
характеризует e-поляризацию молекулы AzOB под влиянием группы N+–O–. Принадлежащая ей ковалентная σ-связь образуется из электрона sp2-орбитали азота и электрона py-орбитали атома O, а ионная связь – за счет переноса второго электрона с sp2-орбитали того же азота на pz-орбиталь кислорода. Принимая на себя второй электрон, pz-орбиталь переходит в состояние $p_{z}^{2}$, а кислород получает отрицательный заряд. В свою очередь, атом N, создавший ковалентно-ионную связь с атомом O и получивший положительный заряд, индуцирует e-поляризацию π-системы, как в схеме 2, за счет локализации обоих π-электронов азосвязи на pz-орбитали азота в группе $\underline {{\text{ - - - }}} {\text{N}}_{ + }^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ → O–. При этом второй азот азогруппы становится катионом ${\text{N}}_{{ \bullet \bullet }}^{ + }$, образующим совместно с сопряженным фенильным кольцом катион PhAT, ответственный за желтый цвет AzOB.
Механизм перегруппировки Валлаха
Механизм процесса ПВ представлен на схеме:

Согласно схеме 4, процесс ПВ начинается с обратимой нейтрализации протоном отрицательного заряда в группе N+–O– (структура A). При этом один из электронов аниона O– переходит с его p2-орбитали на 1s-орбиталь протона и образуется химическая связь из 1s-электрона атома H и оставшегося p-электрона атома O. А возникающая на некоторое время группа OH стабилизирует поляризацию π-системы катиона PhAT (схема 4, A).
Следует отметить, что валентные L-оболочки аниона O– и кислорода группы OH, включив в себя по восемь электронов (схема 4, AzOB, А), приобретают характер L-оболочки инертного неона (2s22p6). Такая ситуация, соответствующая правилу октета Ленгмюра и Косселя [14, с. 62; 15, с. 163], исключает e-пробелы на кислороде и, соответственно, возможность образования химических связей с участием электронов sp2-орбитали соседнего атома ${{{\text{N}}}_{{ \bullet \bullet }}}$. Тем самым, исключается возможность образования рассматривавшихся в [2] треугольных структур с химическими связями между атомом O и азотами азогруппы.
В то же время структура A может присоединить протон по атому ${\text{N}}_{{ \bullet \bullet }}^{ + }$, обладающему неподеленной парой электронов (схема 3, реакция с k2). Эта реакция ведет к переносу заряда на противоположное фенильное кольцо и образованию протонированного азобензола AB$_{1}^{ + }$H одновременно с вытеснением из A гидроксильного катиона HO+. Как отмечалось, для осуществления ПВ используют, как правило, серную кислоту с концентрацией больше 70% [2]. Необходимость такой высокой кислотности следует из схемы 4, согласно которой, протонирование интермедиата A по азоту ${\text{N}}_{{ \bullet \bullet }}^{ + }$ (с константой k2) затруднено из-за кулоновского отталкивания между катионами H+ и ${\text{N}}_{{ \bullet \bullet }}^{ + }$, и естественно полагать, что скорость этой реакции будет меньше, чем обратной реакции диссоциации интермедиата A: k–1[A] > k2[A][H+].
Важным последствием реакции с k2 (схема 4) является не только образование гидроксил-катиона HO+, но и связанной с катионом AB$_{1}^{ + }$H обратимой переполяризации фенильных колец по реакциям с k3 и k–3: AB$_{1}^{ + }$H + H+ ↔ AB$_{2}^{ + }$H + H+. Благодаря этому обеспечивается последовательная замена нейтральных колец (принадлежащих AB$_{1}^{ + }$H и AB$_{2}^{ + }$H), способных реагировать с катионоидными интермедиатами (в актах k5 и k6). Что касается заряженных колец этих изомеров, то для них можно было бы предполагать в качестве реагентов-партнеров анионы HOSO$_{3}^{ - }$, рассматривавшиеся в литературе в качестве активного участника ПВ [2]. Однако характерная для сернокислых растворов AB+H в 98%-ной H2SO4 та же обратимая реакция AB$_{1}^{ + }$H + H+ ↔ AB$_{2}^{ + }$H + H+ с чередованием нейтральных колец не приводит к образованию гидроксиазобензолов при нейтрализации кислотной смеси [5]. Это позволяет принять, что и в условиях ПВ вклад реакции анионов HOSO$_{3}^{ - }$ с фенильными группами AzOB пренебрежимо низок.
Таким образом, в качестве гидроксилирующих агентов AzOB могут выступать только катионы, но не анионы HO– или HOSO$_{3}^{ - }$. При этом важно, что роль гидроксил-катиона HO+, как непосредственного агента гидроксилирования, должна быть небольшой. Такое заключение следует из результата исследования ПВ с препаратом AzOB, содержащим изотоп 18O [2, ссылка 1]. Как оказалось, среди продуктов ПВ выход AB18OH составляет ≈10%, тогда как 90% ABOH не содержат данного изотопа. Отсюда следует, что катионы HO+ преимущественно реагируют с серной кислотой с образованием тех катионов, которые являются непосредственными агентами гидроксилирования.
Согласно схеме 4, непосредственными агентами гидроксилирования должны быть гидроксисульфат-катионы (акты с k5, k6). В ней учтено, что серная кислота в концентрации 72–98% существует в виде комплексов с водой: сульфатов и гидросульфатов гидроксония, причем последние (H3O+ –OSO2OH) доминируют, начиная с 84% H2SO4 [16]. В связи с этим в схеме 4 показано, что реакция HO+ + H3O+ –OSO2OH (с константой k4) приводит к замещению гидроксония и образованию кислоты Каро (HOOSO2OH). В свою очередь, молекулы кислоты Каро тоже взаимодействуют с катионами гидроксония, в результате чего образуются гидроксисульфат-катионы HOSO$_{3}^{ + }$, непосредственно ответственные за гидроксилирование AzOB (акты с k5, k6). Катионам HOSO$_{3}^{ + }$ легче атаковать не положительно заряженные кольца катионов Ph+N+-, а нейтральные фенильные кольца интермедиатов AB$_{1}^{ + }$H и AB$_{2}^{ + }$H. Присоединяясь к атомам углерода в пара-положениях колец с образованием промежуточных σ-комплексов, катионы HOSO$_{3}^{ + }$ вытесняют катионы H+. Вероятно, громоздким катионам HOSO$_{3}^{ + }$ менее выгодно вести атаку по орто-углеродам нейтральных колец вследствие кулоновского противодейстия со стороны группировок ${\text{N}}_{ + }^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$(OH) и ${\text{N}}_{ + }^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$ (схема 4) и необходимости промежуточного изменения геометрии связей атакуемого углерода из плоской в тетраэдрическую. Конечными продуктами ПВ, согласно схеме 4, являются катионы азония с гидроксисульфатными группами в пара-положениях фенильных колец. Их гидролиз при последующей нейтрализации системы приводит к гидроксиазобензолам AB1OH и AB2OH.
Реакции схемы 4 позволяют записать систему дифференциальных кинетических уравнений, которые можно привести к алгебраическим уравнениям. Так, для сопряженных с интермедиатом AB$_{1}^{ + }$H реакций, имеем:
(I)
$\begin{gathered} d[{\text{A}}]{\text{/}}dt = {{k}_{1}}[{\text{AzOB}}][{{{\text{H}}}^{{\text{ + }}}}] - \hfill \\ - \;{{k}_{--}}_{1}[{\text{A}}]--{{k}_{2}}[{\text{A}}][{{{\text{H}}}^{{\text{ + }}}}] = 0, \hfill \\ \end{gathered} $(II)
$\begin{gathered} d[{\text{AB}}_{1}^{ + }{\text{H}}]{\text{/}}dt = {{k}_{2}}[{\text{A}}][{{{\text{H}}}^{ + }}]--{{k}_{3}}[{{{\text{H}}}^{ + }}][{\text{AB}}_{1}^{ + }{\text{H}}] + \\ + \;{{k}_{--}}_{3}[{{{\text{H}}}^{ + }}][{\text{AB}}_{2}^{ + }{\text{H}}]--{{k}_{5}}[{\text{AB}}_{1}^{ + }{\text{H}}][{\text{HOSO}}_{3}^{ + }] = 0, \\ \end{gathered} $(III)
$\begin{gathered} d[{\text{AB}}_{2}^{ + }{\text{H}}]{\text{/}}dt\, = \,{{k}_{3}}[{{{\text{H}}}^{ + }}][{\text{AB}}_{1}^{ + }{\text{H}}]\,--\,{{k}_{--}}_{3}[{{{\text{H}}}^{ + }}][{\text{AB}}_{2}^{ + }{\text{H}}]\,-- \\ - \;{{k}_{6}}[{\text{AB}}_{2}^{ + }{\text{H}}][{\text{HOSO}}_{3}^{ + }] = 0. \\ \end{gathered} $(IV)
$\begin{gathered} d[{\text{A}}]{\text{/}}dt = {{k}_{1}}[{\text{AzOB}}][{{{\text{H}}}^{ + }}]--{{k}_{--}}_{1}[{\text{A}}] \approx 0, \\ {\text{так}}\;{\text{что}}\quad [{\text{A}}] = {{k}_{1}}[{\text{AzOB}}][{{{\text{H}}}^{ + }}]{\text{/}}{{k}_{--}}_{1}. \\ \end{gathered} $(V)
$\begin{gathered} W = {{k}_{5}}[{\text{AB}}_{1}^{ + }{\text{H}}][{\text{HOSO}}_{3}^{ + }] + {{k}_{6}}[{\text{AB}}_{2}^{ + }{\text{H}}][{\text{HOSO}}_{3}^{ + }] = \\ = {{k}_{2}}[{\text{A}}][{{{\text{H}}}^{ + }}] = {{k}_{2}}{{k}_{1}}[{\text{AzOB}}]{{[{{{\text{H}}}^{ + }}]}^{2}}{\text{/}}{{k}_{ - }}_{1}. \\ \end{gathered} $Таким образом, теоретическое выражение скорости образования протонированных гидроксисульфатов AB+H–OSO2OH и получаемых из них путем нейтрализации гидроксиазобензолов согласуется с экспериментально установленной [2] квадратичной зависимостью от концентрации протонов (W ~ [AzOB][H+]2) . При этом именно учтенная в схеме 4 обратимая изомеризация катионов протонированного AB (AB$_{1}^{ + }$H ↔ AB$_{2}^{ + }$H) раскрывает причину образования изомерных гидроксиазобензолов в ПВ.
Отметим, что в схеме 4 не указан второстепенный путь ПВ с низким выходом продуктов перемещения собственного кислорода AzOB в фенильные кольца (≈10%), установленный на меченом Az18OB [2, ccылка 1]. Его можно связать с тем, что небольшая часть образующихся по схеме 4 гидроксилкатионов HO+ все же успевает, не реагируя с серной кислотой, взаимодействовать непосредственно с нейтральными фенильными кольцами интермедиатов AB$_{1}^{ + }$H, AB$_{2}^{ + }$H и, возможно, самого AzOB.
Изложенная выше схема ПВ раскрывает оставшийся без объяснения в [2] путь попадания атома O из кислотной среды в гидроксиазобензолы. Она же позволяет объяснить происхождение таких побочных продуктов, к которым в [2] отнесены незамещенный AB и производное азобензола HOO2SphN=NphOH (4-гидрокси-азобензол-4'‑сульфокислота). Их появление обусловлено реакцией катионов HOSO$_{3}^{ + }$ с серной кислотой конкурентно к актам с константами k5 и k6 (схема 4). Эта реакция уменьшает долю катионов HOSO$_{3}^{ + }$, ответственных за гидроксилирование, а катионы AB$_{1}^{ + }$H и AB$_{2}^{ + }$H, не вступившие в гидроксилирование, превращаются в AB при нейтрализации системы.
Конкурентную для ПВ потерю катионов HOSO$_{3}^{ + }$ можно связать с реакцией: HOSO$_{3}^{ + }$ + H3O+ –OSO2OH → HOO2SOOSO2OH + H3O+, в которой идет замещение катиона гидроксония и образуется пероксодисерная (надсерная) кислота. Пероксодисерная кислота способна распадаться при T ≥ ≥ 65°С на серную кислоту, SO3 и O2 [16, 17], а сульфотриоксид присоединяет протон с образованием катиона сульфония HOO2S+. Образующиеся таким образом катионы HOO2S+ сульфируют нейтральные ph-группы AzOB, AB$_{1}^{ + }$H и AB$_{2}^{ + }$H, приводя в итоге к побочному продукту реакции HOO2SphN=NphOH.
На основании схемы 4, удачно раскрывающей механизм ПВ и природу ответственного за ПВ катиона – HOSO$_{3}^{ + }$, можно объяснить также и отмеченные в [2] “аномалии“ перегруппировок алкильных производных AzOB.
Брутто-схема реакции серной кислоты с 4-алкилазоксибензолами [2]:
(в ней сохранены авторские числовые обозначения) демонстрирует образование обычных продуктов перегруппировки Валлаха (XXXIII), а также отнесенных к “аномальным“ соединений (XXXI), (XXXII, с очень низким выходом), (XXXIV) и алкилазобензолов (XXXV). Отмечается [2], что в случае заместителя X = CH3 преобладает соединение (XXXI), тогда как при X = CH(CH3)2 и C(CH3)3 получаются исключительно соединения (XXXIV). Это, согласно [2], отражает повышенную способность групп CH(CH3)2 и C(CH3)3 вытесняться в виде катионоидных частиц под действием анионов HOO2SO–.
Между тем, выше отмечалось, что в крепкой серной кислоте анионы HO– и HOO2SO– не играют заметной роли в превращениях AzOB. В то же время реально участие катионов HOSO$_{3}^{ + }$. Так, заместитель X = СH3 направляет атаку катиона на орто-позиции фенильного кольца [14, c. 347; 18, с. 301] с образованием σ-комплекса, распадающегося на H+ и гидросульфатный эфир, дающий при гидролизе соединение (XXXI). При этом вытеснение карбокатиона +СН3 катионом HOSO$_{3}^{ + }$ может идти лишь с низким выходом по причине низкой устойчивости этого карбокатиона. В отличие от этого, заместители X = CH(CH3)2 и C(CH3)3 образуют более устойчивые карбокатионы и обеспечивают необходимую доступность катионам HOSO$_{3}^{ + }$ для атаки того углерода кольца, к которому сами прикреплены. Возникающие при этом σ-комплексы диссоциируют на карбокатионы и гидроксисульфатные производные AzOB, являющиеся источником соединений XXXIV при нейтрализации реакционной смеси.
Появление соединения XXXII с низким выходом можно объяснить тем, что вытесняемые катионами HOSO$_{3}^{ + }$ (с низким выходом) карбокатионы +СН3 тоже способны замещать протоны с образованием промежуточных σ-комплексов по орто-углеродам колец, испытывая ориентирующее действие со стороны занявших их место гидроксисульфатных групп.
В обзоре [2] отмечено также, что в реакции H2SO4 с 4-, 4'-диалкил-AzOB (X-ph-ONN-ph-X, где X = CH3, CH(CH3)2 и C(CH3)3) фенильное кольцо группы X-ph-ONN- не изменяется, а в замещении участвует второе кольцо. Теперь при X = CH3 главный продукт тоже получает HO-группу в орто-положении к CH3 вследствие замещения протона катионом HOSO$_{3}^{ + }$. При X = CH(CH3)2 и C(CH3)3 главными продуктами являются Xph-N=Nph-OH и азобензолы Xph-N=Nph-X, а соединения с HO-группой в орто-положении к X имеют лишь следовые количества. Подчеркивается [2], что и в этой реакции тоже проявляется способность алкильной группы образовывать стабильные карбокатионы. Таким образом, и закономерности реакций 4-, 4'-диалкил-AzOB можно связать с участием ключевого катиона HOSO$_{3}^{ + }$.
Следует отметить, что на основе схемы 4 можно восстановить механизм реакции серной кислоты c 2,2',4,4',6,6'-гексаметилазоксибензолом, не способным вступать в перегруппировку Валлаха [2]. Продуктом этой реакции является 4-(гидроксиметил)-2,2',4',6,6'-пентаметилазобензол (выход 91.5% в 85.6%-ной серной кислоте), стехиометрически ее записывают в виде схемы [2]:
В качестве детального механизма этой реакции в [2] была предложена схема с участием анионов –OSO2OH, реагирующих с промежуточными N+≡N+-дикатионами и хиноидными соединениями. Между тем, участие анионов, дикатионов и хиноидных соединений не согласуется с реальной e-формулой азоксибензолов и схемой 4. Теперь естественно полагать, что указанное производное бензилового спирта образуется с участием свободных радикалов, генерируемых термическим разложением промежуточной пероксодисерной кислоты [17]: HOO2SOOSO2OH → 2HOSO2O$ \bullet $.
Реакции радикального замещения водорода в метильной группе могут доминировать над указанным в схеме 4 присоединением катиона HOSO$_{3}^{ + }$ к фенильным кольцам AzOB и катионов AB+H при наличии геометрических препятствий. Последние могут возникнуть от накопленных в молекуле AzOB метильных групп и необходимого для образования промежуточного σ-комплекса (схема 4) изменения валентного состояния углерода кольца (sp2 → sp3) и геометрии его связей от плоской к тетраэдрической [18, с. 298]. Другими словами, строение молекул 2,2',4,4',6,6'-гексаметилазоксибензола неблагоприятно для реакции с катионами HOSO$_{3}^{ + }$, но открывает возможность для реакций свободных радикалов, часть которых отщепляет атомы водорода от метильных групп, а другая часть рекомбинирует с образующимися производными бензильных радикалов. При этом наиболее удобные стерические условия существуют для реакций метильных групп, которые находятся в пара-положениях фенильных колец, а образующиеся с ними сульфатные эфиры переходят в ходе нейтрализации кислой среды в 4-(оксиметил)-2,2',4',6,6'-пентаметилазобензол.
В заключение следует отметить, что интерпретация экспериментальных данных по перегруппировке Валлаха до сих пор базировалась на неадекватном понимании формулы строения азоксибензола. Это не позволяло установить реального механизма перегруппировки. Между тем, такой механизм получается с учетом характера e-поляризации AzOB и участия в ПВ таких интермедиатов, как гидроксисульфат-катионы HOSO$_{3}^{ + }$ и их предшественники гидроксил-катионы HO+. При этом некорректная идея о роли гидроксисульфат-анионов –OSO2OH заменяется естественым механизмом электрофильного замещения. А при невозможности такового для некоторых производных AzOB, как в случае с 2,2',4,4',6,6'-гексаметилазоксибензолом, могут идти реакции с участием свободных радикалов.
Список литературы
Химическая энциклопедия / Под ред. И.Л. Кнунянца. Т. 1. М.: Сов. Энциклопедия, 1988. 347 с.
Фурин Г.Г. // Успехи химии. 1987. Т. 56. № 6. С. 911.
Cox R.A. // J. Am. Chem. Soc. 1974. V. 96. № 4. P. 1059.
Курс физической химии. Т. 1 / Под ред. Я.И. Герасимова. М.-Л.: Химия, 1964. С. 165.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 2. С. 243.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2017. Т. 91. № 10. С. 1683. https://doi.org/10.1134/S0036024417090199
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 2. С. 267. https://doi.org/10.1134/S0036024418020164
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 10. С. 1552. https://doi.org/10.1134/S0036024418100205
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 2. С. 313. https://doi.org/10.1134/S0036024419010205
Михеев Ю.А., Ершов Ю.А. // Там же. 2019. Т. 93. № 7. С. 1111. https://doi.org/10.1134/S0036024419070185
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94. № 1. С. 143. https://doi.org/10.1134/S0036024420010227
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94. № 8. С. 1269. https://doi.org/10.1134/S0036024420080208
Михеев Ю.А., Ершов Ю.А. // Там же. 2020. Т. 94, № 11. С. 1706. https://doi.org/10.1134/S0036024420110254
Темникова Т.И. Курс теоретических основ органической химии. Л.: Гос. научно-технич. изд-во хим. лит-ры, 1962. 948 с.
Коулсон Ч. Валентность. М.: Мир. 1965. С. 141.
Некрасов Б.В. Основы общей химии. Т. 1. М.: Химия, 1973. С. 338.
Яндекс. ru.wikipedia.org. Пероксодисерная кислота.
Стрейтвизер Э. Теория молекулярных орбит для химиков-органиков. М.: Мир, 1965. С. 296.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии