

Журнал физической химии, 2022, T. 96, № 10, стр. 1390-1403
Физико-химические механизмы и кинетические законы, определяющие экстремальные скорости реакций в горении и детонации газов
В. В. Азатян a, *, В. М. Прокопенко b, **, Т. Р. Тимербулатов c
a НИИ системных исследований РАН
Москва, Россия
b Институт структурной макрокинетики и проблем материаловедения им. А.Г. Мержанова РАН
Черноголовка, Московская область, Россия
c Группа компаний КОНТИ
Москва, Россия
* E-mail: vylenazatyan@yandex.ru
** E-mail: nelly.prokopenko@yandex.ru
Поступила в редакцию 13.04.2022
После доработки 13.04.2022
Принята к публикации 20.04.2022
- EDN: WZKAKG
- DOI: 10.31857/S0044453722100053
Аннотация
Обобщены результаты исследований кинетических особенностей реакций в горении, взрыве и детонации газов с комплексным использованием кинетических и спектроскопических методов. Установлено, что, вопреки прежним представлениям, во всех режимах горения химические процессы, являются цепными реакциями и протекают по ранее неизвестным законам неизотермических цепных процессов. Показано, что в распространяющемся пламени и в детонации характеристические времена реакции меньше соответственно десятитысячной и миллионной доли секунды. Выяснены кинетический механизм и закон, управляющий такими скоростями и ускорениями, а также их чрезвычайно сильной температурной зависимостью. Показана решающая роль высоких концентраций атомов и радикалов. Разработаны эффективные химические методы управления всеми режимами горения.
Повышенный интерес к горению, взрыву, детонации газов определяется стратегической ролью этих процессов в технике, в энергетике и в быту, а также специфическими закономерностями реакций, относящимися к актуальным проблемам теорий химической кинетики и горения. На основе многолетних исследований разработана теория газодинамики этих процессов. Фундаментальные же проблемы химических и физико-химических аспектов явлений начали получать свое решение лишь в последние два–три десятилетия. Настоящий обзор посвящен кардинальной проблеме физико-химических механизмов и кинетических законов, определяющих особенности крайне быстрых реакций в процессах горения, взрыва и детонации газов.
Горение – химический процесс, протекающий на стадиях развития c прогрессирующим самоускорением и сопровождающийся выделением тепла и света. Возгорание, распространение пламени, взрыв и детонация это – различные режимы горения. В газовой фазе протекает горение не только соединений, газообразных в обычных условиях, но и многих конденсированных веществ, которые перед возгоранием испаряются. Газофазное горение в зависимости от химического механизма и условий может быть вызвано или прогрессивно усиливающимся саморазогревом реакционной смеси или лавинным размножением активных промежуточных продуктов, которое при интенсивном протекании также сопровождается саморазогревом. Горение, вызванное только саморазогревом, называют тепловым.
В работах [1, 2] было показано, что тепловое воспламенение происходит, если скорость тепловыделения (q+) больше, чем скорость теплоотвода (q_), и если при этом от повышения температуры (Т) тепловыделение ускоряется больше, чем теплоотвод:
Скорость тепловыделения, равна произведению скорости реакции (W) и теплового эффекта ($\bar {Q}$): Скорость теплоотвода выражают уравнением: где S и V – величины поверхности и объема реакционной камеры, $\alpha $ – коэффициент теплоотдачи, T0 – температура стенок реактора.Соотношение (1) является условием саморазогрева. Соотношение же (2) соответствует режиму, в котором от повышения температуры тепловыделение ускоряется больше, чем теплоотвод. Одновременное выполнение условий (1) и (2) приводит к прогрессирующему накоплению тепла в системе и к возрастающему ускорению процесса. Теорию, рассматривающую саморазогрев как единственный фактор самоускорения реакции, называют тепловой теорией горения.
1. ЦЕПНОЕ ВОСПЛАМЕНЕНИЕ
В отличие от модели теплового горения, содержащей только реакцию валентно-насыщенных молекул, цепное горение протекает при определяющем участии свободных атомов и радикалов, которые в реакциях с исходными реагентами регенерируются. Многократно чередующиеся такие реакции образуют реакционную цепь. В определенном типе реакций происходит размножение этих активных частиц и, соответственно, ускорение их реакций с исходными реагентами [2–4]. Такого типа процессам соответствует следующая схема:
где x и y – свободные атомы и радикалы – носители цепей (НЦ), A и B – исходные молекулы, Р – конечный продукт.При наличии свободных валентностей у свободных атомов и радикалов энергии активации их реакций с валентно-насыщенными молекулами на десятки ккал/моль меньше, чем энергии активации межмолекулярных реакций. Соответственно, константы скорости реакций НЦ в тысячи раз больше, и цепное горение протекает даже при давлениях в сотни раз ниже атмосферного. Особенности цепного горения ниже рассматриваются на примере модельной реакции горения водорода. Реакционная цепь состоит из многократно повторяющейся совокупности элементарных реакций (I), (II) и (III) [2–4], образующих звено реакционной цепи:
| Н2 + О2 = 2OH или/и | ∆H | E | |
| Н2 + О2 = Н + НО2 | 238.0 | 225.0 | (0) |
| Н + О2 = ОН + О | 68.6 | 70.0 | (I) |
| ОН + Н2 = Н2О + Н, | –58.6 | 21.9 | (II) |
| О + Н2 = ОН + Н | 8.4 | 44.4 | (III) |
| Н + стенка → обрыв. | (IV) |
Справа приведены изменения энтальпии и энергии активации в кДж/моль [5, 6]. Лимитирующей стадией в звене является реакция (I), без которой периодическая регенерация НЦ невозможна. Эта стадия медленнее остальных двух стадий звена. В стадии (I) происходит также размножение свободных валентностей, обеспечивающее разветвление цепей. В силу большой величины константы скорости стадии (II) в эту реакцию быстро вступают радикалы ОН, образованные как в стадии (I), так и в стадии (III). Таким образом, звено цепи завершается регенерацией и размножением атомарного водорода и выделением всего 46 кДж/моль:
(A)
${\text{Н}} + 2{{{\text{Н}}}_{2}} + {{{\text{О}}}_{2}} + {{{\text{Н}}}_{2}} = 2{{{\text{Н}}}_{{\text{2}}}}{\text{О}} + 3{\text{Н}}.$(VI)
${\text{Н}}{{{\text{О}}}_{2}} + {{{\text{Н}}}_{2}} = {{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{2}} + {\text{Н}}$Атомы Н погибают также при хемосорбции на стенках и в реакции с ингибитором (In):
Для рассмотрения закономерностей цепного горения система кинетических уравнений активных частиц методом частичных квазистационарных концентраций [3] приводится к одному уравнению, относящемуся к тому НЦ, реакция которого осуществляет разветвление и лимитирует процесс. В данном процессе, как было указано, этот НЦ – атом Н. Обозначая для общности О2, через В, и концентрацию атомов Н через n, на основании реакций (0)–(IV) изменение концентрации носителей цепей n выражают следующим уравнением [2, 3]:
где f и $g$ – скорости реакций (I) и (IV) НЦ при их единичных концентрациях, в которых эти частицы размножаются или погибают соответственно. Эти скорости равны:(6)
$\begin{gathered} f = 2{{k}_{1}}[{\text{B}}] = 2k_{1}^{{\text{o}}}{{e}^{{--~\frac{{{{E}_{1}}}}{{RT}}}}}[{\text{B}}], \\ g = {{k}_{{{\text{эфф}}}}}, \\ \end{gathered} $Скорости же расходования О2, т.е. скорости процесса, соответствует уравнение:
(7)
$d[{\text{B]/}}dt = {{\omega }_{0}} + \left\{ {{{k}_{1}} + ~\,\,{{k}_{5}}[{\text{M}}]} \right\}[{\text{В}}]{\kern 1pt} n = \,~\,{{\omega }_{0}} + \,\,~{{k}_{c}}[{\text{В}}]{\kern 1pt} n,$Роль реакций обрыва цепей с участием активных частиц ОН и О незначительна, в силу очень больших величин констант скорости стадий (II) и (III), в которых эти НЦ регенерируют. Из уравнения (5) следует, что в условиях, при которых f < $g$, устанавливается очень низкая стационарная концентрация активных частиц, определяемая соотношением скоростей их образования в очень медленной реакции (0) и гибели. Если же f > $g$, т.е. если размножение НЦ быстрее их гибели, то происходит лавинное размножение активных частиц. Соответственно, лавинообразно ускоряется и расходование исходных реагентов, реагирующих с этими НЦ в реакциях (I)–(III). Таким образом, условием цепного воспламенения является неравенство:
Cаморазогрев сильно ускоряет реакцию горения. Это оказалось основной причиной того, что было общепринятым считать горение при наличии саморазогрева не цепным, а тепловым. При этом химический процесс горения отождествляли с одностадийной реакцией между исходными молекулами (например, [7–21]). Для аналитического описания процесс произвольно считали одностадийной реакцией. По измеренной скорости и с использованием также произвольно принятого уравнения реакции первого (реже второго) кинетического порядка определяли “константу скорости”. Таким способом удавалось описать (не объяснить) лишь отдельные характеристики кинетики в узких условиях, относящихся к расчетам. Без учета реакций свободных атомов и радикалов рассматривались распространение пламени и детонация (например, [7–22]). С использованием нереальных констант скорости нередко проводилось также численное моделирование (например, [15, 16]). В работе [16] процессы горения 2Н2 с О2 и 2СО с О2 представлены как одностадийные тримолекулярные реакции, которым приписаны константы скорости реакций второго кинетического порядка с явным нарушением физического смысла и размерности. Приписываемые этим реакциям скорости завышены во много тысяч раз. Ясно, что такие работы противоречат самому факту горения и законам химической кинетики.
В серии теоретических и экспериментальных исследований (например, [23–26]), было установлено, что модель одностадийной реакции противоречит факту реального горения. В силу энергий активации, превышающих 200 кДж/моль, реакции между валентно-насыщенными молекулами в тысячи раз медленнее реальных реакций горения и практически даже не сопровождаются саморазогревом (см. ниже). В работах [23–28] было установлено также, что, вопреки общепринятым представлениям, горение газов протекает по цепным механизмам не только при давлениях в десятки и сотни раз ниже атмосферного, но и при любых давлениях и температурных режимах. Во всех режимах горения реакции протекают по не известным ранее законам неизотермических цепных реакций, проявляя особые закономерности.
Настоящий обзор посвящен проблеме физико-химического механизма чрезвычайно быстрых реакций горения, взрыва и детонации соединений вопреки их очень прочным химическим связам. Рассмотрена специфика выявленных законов неизотермических цепных реакций.
2. ЦЕПНАЯ ПРИРОДА ГОРЕНИЯ, ВЗРЫВА И ДЕТОНАЦИИ ГАЗОВ
Доказательство цепного характера реакций горения и их закономерности ниже описываются на примере модельной реакции горения водорода. Был разработан метод доказательства, заключающейся в том, что малые присадки выбранных нами ингибиторов-олефинов, обрывают реакционные цепи, блокируют цепную лавину, и горение становится невозможным, несмотря на инициирование искрой, раскаленной проволокой или ударной волной даже при высоких температурах. Олефины – молекулы с двойными связями, намного эффективнее широко используемых в пожаротушении хладонов. Воспламенение регистрировалось по появлению пиков на осциллограммах хемилюминесценции и давления, а также визуально и по звуковому эффекту.
Из рис. 1 видно что, например, 1.5% изобутена предотвращают воспламенение всех водородо-воздушных смесей, содержащих более 35% Н2. Видно также, что в отличие от наших ингибиторов, для предотвращения горения требуются количества N2 и СО2, превышающие 60 и 50%, т.е. требуется сильное разбавление смеси. Показано, что возгорание предотвращается в результате очень быстрой реакции замены атомов Н на радикал i-С4Н9:
(VIII)
${\text{Н}} + i{\text{ - }}{{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{{\text{8}}}} = i{\text{ - }}{{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{9}},$Рис. 1.
Влияние ингибиторов: изобутена (1), пропилена и гексена-2 (2), СО2 (3) и азота (4) на концентрационные пределы.

То, что после запуска искры смесь остается неизменной, означает, что исходные реагенты между собой практически не реагируют. Аналогичным способом показано предотвращение горения метана, пропана и синтез-газа, паров керосина [27]. Таким образом, эксперименты подтверждают вывод о протекании воспламенения только по цепному механизму, показывают, что скорости межмолекулярных реакций в тысячи раз меньше скорости реакций горения.
Доказательства цепной природы воспламенения при высоких температурах, а также перехода горения во взрыв и в детонацию, были получены с помощью разработанных нами методов, использующих ударные волны (УВ) [25–27], а также явление ингибирования. Методы позволяют повысить температуру газа за доли микросекунды на тысячи градусов, инициировать горение при заданных температурах (до 1700 К) и следить за влиянием ингибиторов на горение при заданных начальных температурах, однородных по объему реактора. Было показано, что даже при высоких начальных температурах и при очень сильном инициировании благодаря ингибиторам, обрывающим реакционные цепи, воспламенение и горение предотвращаются. Например, из рис. 2 видно, что смесь, воспламеняющаяся в отсутствие ингибиторов при 930 К и 60 кПа, не удается воспламенить даже при 1130 К и давлении 84 кПа (0.83 атм) в результате подавления цепной лавины присадкой 0.7% пропилена: не регистрируются ни свечение, ни кратковременный рост давления, присущие воспламенению. Для воспламенения смеси с такой примесью приходится увеличить скорость инициирующей УВ до 2.74 км/с и, тем самым, повысить температуру и давление до 1140 К и 0.85 атм соответственно. При дальнейшем увеличении содержания ингибитора минимальная температура воспламенения и критическое давление становятся еще выше (рис. 2). Таким образом, действительно, даже при указанных высоких температурах молекулярные реагенты непосредственно между собой не реагируют. Уже из этих данных следует, что, поскольку послойное воспламенение служит начальной стадией распространения пламени и детонации, то реакции в указанных режимах горения также являются цепными.
Рис. 2.
Влияние пропилена (1) и паров изопропанола (2) на минимальную температуру воспламенения смесей водорода с воздухом; 3 и 4 – температуры воспламенения пропилена и изопропанола в воздухе.

Резкое ускорение реакций во взрыве тепловая теория горения объясняет сильным повышением температуры при адиабатическом сжатии. Приведенные ниже результаты, однако показывают, что основную роль играет неизвестный ранее особый закон температурной зависимости скорости цепной реакции рассматриваемый в разделе 4.
3. ОСОБЕННОСТИ ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ СКОРОСТИ РЕАКЦИЙ ГОРЕНИЯ
Зависимость скорости реакции от температуры определяет важнейшие характеристики горения, в том числе условия перехода во взрыв и в детонацию. Между тем, основные положения теории горения и ее методов до последнего времени основывались на ряде неправильных представлений о температурной зависимости скорости реакций горения. Это влияло на достоверность многих выводов учения о горении (см. ниже). Было общепринятым считать, что температурная зависимость скорости соответствует закону Аррениуса [3, 7–22, 29–31], и что зависимости скорости и ее константы от температуры тем сильнее, чем больше энергия активации [1, 7, 10, 12, 22]. Оказалось, однако, [27, 32–34], что эти представления неправильны и противоречат экспериментам.
Поскольку температурные зависимости концентраций и констант скорости разные, то в ходе горения эти величины изменяются по качественно разным законам. Даже в простейшем случае первого кинетического порядка скорость реакции $W(T,t)$ равна произведению двух разных функций от температуры: константы скорости $k(T,t)$ и концентрации $C(T,t)$:
Из уравнения (9) видно, что изменение скорости при изменении температуры, равно:
(10)
$\frac{{\partial W}}{{\partial {\text{T}}}} = k(T,t)\frac{{\partial C(T,t)}}{{\partial T}} + C(T,t)\frac{{\partial k(T,t)}}{{\partial T}}.$Встречающееся часто отождествление температурных зависимостей $W(T,t)$ и $k(T,t)$ – неоправданное игнорирование второго слагаемого в уравнении (10). Это – одна из основных причин того, что большие скорости реакций горения газов и их температурные зависимости, в том числе, в режимах взрыва детонации, до недавнего времени не находили объяснения.
Коренное различие температурных зависимостей $W(T,t)$ и $C(T,t)$ видно на примере уравнения (9), интегрирование которого при начальной концентрации $~{{С}_{0}}$ дает
(11)
${\text{С}}(T,t) = {{C}_{0}}\exp \left[ {--\mathop \smallint \limits_{{{t}_{0}}}^t \,k(T,t)} \right]dt,$(12)
$W(T,t) = k(T,t){{C}_{0}}\exp \left[ {~--\mathop \smallint \limits_{{{t}_{0}}}^t \,k(T,t)} \right]dt.$Неправильны также представления, согласно которым, более сильным температурным зависимостям W(T) и k(T) якобы соответствуют большие энергии активации. Действительно, мерой температурной зависимости константы скорости служит абсолютная величина ее изменения при единичном изменении температуры [27, 32]: ${{\beta }} = \frac{{\partial k}}{{\partial T}}$. Этой производной определяется абсолютная величина изменения скорости реакции, вызванного изменением температуры при данных концентрациях реагентов. Именно абсолютная величина изменения скорости, вызванного изменением температуры, определяет величину саморазогрева, переход во взрыв и в детонацию. Из выражения для константы скорости бимолекулярной реакции:
следует, что ее зависимость от температуры определяется выражением:(14)
$\beta = \frac{{dk}}{{dT}} = {{k}^{0}}{{e}^{{--~\frac{E}{{RT}}}}}\frac{E}{{R{{T}^{2}}}} = k\frac{E}{{R{{T}^{2}}}}.$В силу больших энергий активации межмолекулярных реакций (Е = 200–225 кДж/моль) экспоненциальный множитель крайне мал. Поскольку, кроме того, величина k0 не может превышать частоту двойных столкновений, то при большой энергии активации величина $~{{\beta }}$ крайне мала, т.е. зависимость константы скорости от температуры очень слабая. Энергии активации реакций атомов и радикалов намного меньше, и значит, константы скорости намного больше. Поэтому температурная зависимость скоростей элементарных реакций активных частиц намного сильнее. Это различие наглядно иллюстрируют приведенные ниже формулы и данные рис. 3. Видно, что даже при 3500 К крайне мала не только константа скорости межмолекулярной реакции, но и незначителен абсолютный рост ее величины. Константа скорости свободных радикалов так, же как и их рост несравненно больше (кривая 1). Температурная зависимость скорости цепных процессов определяется прежде всего, спецификой их кинетических законов (см. следующий раздел).
Рис. 3.
Графики функции k/kо = ехр(– Е/RT) при различных значениях энергии активации: 1 – 25, 2 – 150, 3 – 225 кДж/моль. На вставке показан участок высоких температур.

Было показано [27, 32, 35] что утверждения о сильной температурной зависимости при больших энергиях активации – следствие того, что вместо абсолютных величин изменений W(T) и k(T) рассматривались относительные величины, т.е. $\frac{{\Delta k}}{k}$ и $\frac{{\Delta W}}{W}$. Между тем, эти дроби кинетику реакции нисколько не определяют. Влияние изменения температуры на скорости определяется не и относительными изменений k(T) и W(T), а абсолютными величинами изменений.
4. ЗАКОН ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ СКОРОСТИ ЦЕПНОГО ГОРЕНИЯ
Большие скорости и ускорения реакций во взрыве тепловая теория горения связывает только с сильным повышением температуры при адиабатическом сжатии. Приведенные ниже результаты, однако показывают, что основную роль играет выявленный нами особый характер температурной зависимости скорости реакции. Принципиальное отличие этой зависимости от аррениусовской функции проявляется, например, в следующем. При нагревании стехиометрической смеси Н2 с воздухом при 1 атм от 823 до 853 K константа скорости межмолекулярной реакции, характеризующейся энергией активации 225 кДж/моль, увеличивается лишь на ≈5%. Так же мало возрастет скорость этой реакции (0). Рост скорости расходования О2 в реакции (0) составляет лишь 0.5%. Между тем, в эксперименте при таком же повышении температуры смесь воспламеняется и выгорает за десятитысячные доли секунды, т.е. ускоряется на несколько порядков больше, чем следовало ожидать по закону Аррениуса. Такая сильная зависимость определяется следующим законом изменения концентрации НЦ. В каждый момент времени и при каждых данных величинах f и g скорость $\frac{{dn}}{{dt}}$ находится в обратной связи с их концентрацией n, как это видно также из уравнения (5). При этом, если f > g, то обратная связь положительная, и из (5) следует, что величина n возрастает во времени экспоненциально, даже при постоянной температуре:
В величину же f входит константа скорости k1 со своим фактором Больцмана в соответствии с выражением (6). Таким образом, при f > g концентрация n и скорость W зависят от температуры по закону экспоненты, находящейся в положительном показателе степени (факторе Больцмана). Очевидно, что такая зависимость несравненно сильнее, чем экспонента в законе Аррениуса. Поскольку эта зависимость осуществляется при каждой данной температуре, то она выполняется в ходе горения.При t > 2.5/φ, когда в уравнениях (5) и (7) уже можно пренебречь величиной ω0, интегрирование уравнения (5) с учетом выражения f по формуле (7) и температурной зависимости k1 приводит к следующей зависимости n от температуры и времени:
(16)
$n = {{n}_{0}}\exp \mathop \smallint \limits_{{{t}_{0}}}^t \left[ {{{f}_{0}}\exp \left( {--\frac{{{{E}_{p}}}}{{RT}}} \right) - g} \right]dt.$При подстановке $n$ из выражения (16) в уравнение скорости (5) получается [27, 32, 33]:
(17)
$\frac{W}{{[{\text{B}}]}} = {{k}_{1}}{{n}_{{0~}}}\exp \mathop \smallint \limits_{t0}^t \left[ {{{f}_{0}}\exp (--{{E}_{p}}{\text{/}}RT)--g} \right]dt.$Мы видим, что подобно концентрации НЦ, скорость реакции зависит от температуры по закону экспоненты, содержащей в положительной экспоненте фактор Больцмана. Из выражения (17), количественно подтвержденного экспериментом [27, 34], следует также, что в развивающемся горении по такому же закону возрастает и величина $\frac{{\partial W}}{{\partial T}},$ т.е. по такому же закону усиливается температурная зависимость скорости. Обратим внимание на то, согласно выражению (17), скорость возрастает также вследствие увеличения времени реакции, находящегося в показателе степени. Именно такая сильная температурная зависимость скорости реакции обусловливает быстрое распространение пламени, легкий переход горения во взрыв (см. раздел 7) и в детонацию, определяет описанные ниже чрезвычайные скорости и самоускорения реакции во всех режимах горения даже в отсутствие адиабатического сжатия. Формула (17) описывает завершение реакции горения водорода за десятитысячные доли секунды, наблюдаемое в эксперименте с распространением пламени. Авторы [7, 22], не учитывавшие цепной характер горения, наблюдаемый резкий рост скорости реакции сравнивают с известной из квантовой механики δ-функцией. Аналогия, однако, чисто формальная и, конечно, δ-функция не объясняет явления.
5. ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА ЗАКОНА ЭКСПОНЕНТЫ ПРИ ПОЛОЖИТЕЛЬНОМ ПОКАЗАТЕЛЕ СТЕПЕНИ (ФАКТОРЕ БОЛЬЦМАНА)
Температурную зависимость скорости реакции горения и концентраций НЦ изучали в широкой области давлений и в интервале температур от 763 до 2500 К. В одной из серий экспериментов горение стехиометрической смеси Н2 с О2 в термостатированном кварцевом реакторе проводилось при постоянных температурах 773 и 768 K и начальном давлении 2.25 Торр. Схема установки приведена на рис. 4. Микротермопара помещалась в кварцевом капилляре, промытом плавиковой кислотой для уменьшения гетерогенной рекомбинации атомов и рекомбинационного разогрева. Из рис. 5 видно, что различие разогревов при заданных начальных температурах не превышает 0.15 К, т.е. меньше 3% от разности начальных температур. Регистрированный же максимальный рост температуры 4 К вызван в основном рекомбинационным разогревом термопары, о чем свидетельствует также то, что максимум разогрева достигается лишь позже максимальной интенсивности хемилюминесценции и максимальной скорости падения давления, т.е. после реакции, вследствие инерционности термопары. Благодаря гетерогенному обрыву цепей, отводящему энергию рекомбинации, низким давлениям и большой скорости кондуктивного теплоотвода обеспечивалась практическая изотермичность процесса. Время достижения максимальной скорости, отсчитанное даже после периода индукции, составляло 0.36–0.5 с, что в тысячи раз превышало время теплоотвода. Поскольку, таким образом, саморазогрев незначителен и практически одинаков при заданных начальных температурах, то при рассмотрении отношения скоростей с большой точностью можно пренебречь его вкладом.
Рис. 4.
Блок-схема установки для кинетических исследований при низких давлениях: 1 – терморегулятор термостата, потенциометрический регистратор температуры, 2 – термопара, 3 – вакуумная установка, 4 – мембранный манометр, 5 – преобразователь давления, 6 – вакуумный пост, 6'–8 – усилители постоянного тока, 9 – фотоумножитель, 10 – многоканальный осциллограф.

Рис. 5
Осциллограммы давления (1, 1'), хемилюминесценции (2, 2') и температуры (3, 3') при 768 (1–3) и 773 K (1'–3').

Данные рис. 5 позволяют сравнивать рост скорости реакции с ростом константы скорости лимитирующей стадии, соответствующим общепринятому допущению о подчинении температурной зависимости скорости закону Аррениуса. Согласно стехиометрии процесса:
уменьшение числа молей смеси равно уменьшению числа молей О2. Поэтому отношение максимальных величин угловых коэффициентов кинетических кривых 1 и 1' равно отношению измеренных максимальных скоростей расходования О2 при 773 и 768 K. Это отношение равно 1.29, т.е. при нагревании на 5 К скорость возрастает на 29%. Между тем, увеличение константы скорости лимитирующей стадии (I) k1 = 1.87 × × 1014exp(–8350/T) см3 моль–1 с–1 [6, 27] при таком повышении температуры составляет лишь 7%. Таким образом, намного более сильная температурная зависимость скорости реакции по сравнению с традиционно используемым законом Аррениуса проявляется даже при разности температур лишь 5 К.Как видно из табл. 1, реальное увеличение скорости, вызванное повышением температуры, находится в хорошем согласии с рассчитанным по уравнению (17). Небольшой рост расчетного отношения скоростей во времени (второй столбец) обусловлен тем, что расходование О2 в расчетах не учитывалось, и, соответственно, расчетная скорость во времени несколько возрастала.
Таблица 1.
Экспериментальное (I) и рассчитанные по формуле (17) и формуле Аррениуса (II и III) отношение $~{{W}_{2}}{\text{/}}{{W}_{1}}$при 773 и 76
| t – t0, c | I | II | II |
|---|---|---|---|
| 0.06 | 1.60 | 1.45 | |
| 0.08 | 1.70 | 1.62 | 1.05 |
| 0.10 | 1.62 | 1.75 |
6. РОЛЬ РАЗМНОЖЕНИЯ АКТИВНЫХ ЧАСТИЦ В ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ СКОРОСТИ РЕАКЦИИ В РАСПРОСТРАНЯЮЩЕМСЯ ПЛАМЕНИ
Различие температурных зависимостей скорости реакции и величины k1 позволяет определить вклад роста концентрации НЦ в наблюдаемую “аномальную” зависимость скорости от температуры. Выразив условно температурную зависимость скорости законом Аррениуса, можно записать известное уравнение:
(18)
${\text{ln}}({{W}_{2}}{\text{/}}{{W}_{1}}) = \frac{{E({{T}_{2}} - {{T}_{1}})}}{{R{{T}_{1}}{{T}_{2}}}},$Подставив отношение тангенсов максимальных углов наклонов осциллограмм на рис. 5, равное 1.29, и значения Т1 и Т2 в это выражение, получаем эффективную величину энергии активации процесса, равную 60.0 ккал/моль. Между тем, энергия активации константы скорости лимитирующего акта (I) равна лишь 16.7 ккал/моль [6, 27]. Из этих данных и из выражения скорости расходования О2:
(19)
$W = --\frac{{d\left[ {{{{\text{O}}}_{2}}} \right]}}{{dt}} = {{k}_{1}}\left[ {\text{H}} \right]\left[ {{{{\text{O}}}_{2}}} \right],$Полученные результаты позволяют оценить концентрацию атомов Н, обеспечивающую наблюдаемую скорость реакции и превышение ее роста над увеличением константы скорости k1. Разделив максимальное значение скорости W на соответствующую ей концентрацию О2, рассчитанную по величине давления (рис. 5), получим концентрацию атомов Н в момент максимальной скорости, равную 1.3 × 1014 атом/см3. Эта величина вместе с приведенной выше энергией активации 43 ккал/моль находится в хорошем согласии с измеренной ранее [36] методом ЭПР концентрацией атомов Н в пламени такой же смеси при 930 К и 3 Торр (~3 × 1015 атомов/см3). Действительно, из приведенных величин концентрации атомов Н и энергии активации получается концентрация при 930 К, равная 2.3 × 1015 атом/см3. Таким образом, вывод о ведущей роли роста концентрации НЦ в температурной зависимости скорости количественно подтверждается результатами измерений W, ΔT и концентрации атомов.
Роль размножения НЦ в росте скорости реакции и ее температурной зависимости еще значительнее при большей разности температур. Ниже сопоставляются скорость при 843 К (температура самовоспламенения около третьего предела при 1 атм в кварцевом реакторе [5]) и скорость при температуре распространения пламени в условиях отсутствия взрыва, т.е. в отсутствие адиабатического сжатия. Скорость реакции, приведенная в монографии [5], равна 7.2 × 1015 молек./(см3с). При такой скорости характеристическое время реакции на три порядка больше времени теплоотвода. Поэтому саморазогрева практически нет.
Распространение пламени проводилось в смеси 15% Н2 с воздухом в молибденовой трубке диаметром 1.2 см и длиной 210 см с открытым концом (рис. 6). Вдоль трубки размещались фотоумножители, сигналы которых передавались в многоканальный осциллограф, соединенный с компьютером. Для устранения помех постороннего света трубка обертывалась черной бумагой. С целью уменьшения гетерогенной рекомбинации трубка споласкивалась борной кислотой. По данным хроматографического анализа, в пламени водород выгорает практически полностью. В соответствии со стехиометрией, О2 расходуется вдвое меньше.
Рис. 6.
Схема установки изучения распространения пламени: 1 – высоковольтный блок питания, 2 – осциллограф, 3 – фотодатчики, 4 – реактор, 5 – кран, 6 – вакууметр, 7 – вакуумный насос, 8 – кран пробоотборника.

Адиабатический разогрев составляет 1070 К. При допущении потерь 15% разогрев равен 910 К. Соответственно температура пламени равна 1203 К. Скорость реакции определяли как отношение концентрации израсходованного О2 ко времени нахождения смеси в зоне пламени. Это время определялось как отношение толщины пламени и ее скорости. Скорость пламени определялась по наклону х–t-диаграмм пробега пламени и составляла ~23 м/с. Ширина зоны пламени (~0.15 см) определялась по известному для данного состава смеси профилю температуры вдоль пламени [37]. Такой ширине при данной скорости пламени соответствует продолжительность реакции <10–4 с.
Среднюю скорость реакции определяли как отношение количества кислорода ко времени нахождения смеси в зоне реакции. Учитывая начальную концентрацию Н2 при этой температуре, равную 2.2 × 1017 молек/см3 с, продолжительность реакции (<10–4 с), скорость реакции горения по водороду получаем большей 2.2 × 1021молек/(см3 с) и, соответственно скорость по кислороду большей 1.1 × 1021молек./(см3 с). Таким образом, экстремальный характер скорости реакции проявляется в том, что менее, чем за десятитысячную долю секунды в каждом см3 расходуются практически все 2.2 × 1017 прочные молекулы Н2 и эквивалентное количество О2 с еще более прочными молекулами.
Другое проявление экстремального характера кинетики реакции заключается в ускорении. При 843 К скорость в стехиометрической смеси Н2 с О2 составляет 7.2 × 1015 молек/(см3 с) [3]. С учетом поправок (различие плотностей газа и различие начальных составов) скорость при 1203 К равна 4.87 × 1022 молек/(см3 с). Это значит, что при повышении температуры с 843 до 1203 К процесс ускоряется в 6.7 × 106 раз. Константа же скорости реакции (I) при таком повышении температуры возрастает лишь в 20 раз, т.е. ускорение по закону Аррениуса в 3.3 × 105 раз меньше реального.
Поскольку О2 расходуется только в реакции с атомарным водородом, то очевидно, что различие между возрастаниями W и k1 вызвано ростом концентрации атомов Н, т.е. скорость и самоускорение реакции при распространении пламени определяются практически только ростом концентрации активных частиц. Поделив скорость на концентрацию О2 и на k1, получаем концентрацию атомов Н в пламени, равную 4.8 × 1015 атомов/см3, что составляет 0.5% от [Н2]. Эта величина близка к измеренной в пламени смесей аналогичного состава методом масс-спектрометрии [37]. Приведенные результаты подтверждают данные об использовании системой основной части энергии реакции для образования атомарного водорода (раздел 1).
Таким образом, скорость пламени, равная 23 м/с, даже в бедной водородом смеси в трубке небольшого диаметра реализуется благодаря экстремальным ускорениям и скоростям реакции, высоким концентрациям атомов Н, размножающихся по закону экспоненты с положительным показателем степени (фактор Больцмана) в соответствии с уравнением (17).
7. ЦЕПНОЙ ХАРАКТЕР РЕАКЦИЙ ВО ВЗРЫВЕ
При одновременном выполнении условий цепной и тепловой лавины, т.е. соотношений (8), (1) и (2), реакция в силу закона (17) экспоненты с положительным показателем степени (фактор Больцмана) ускоряется настолько резко, что ее характеристическое время становится короче времени расширения давления в ответ на рост температуры на расстояниях, превышающих размеры очага реакции. При таких скоростях реакции даже в области давлений в сотни раз ниже атмосферного давления возникает скачок скорости реакции и температуры [27, 38, 39]. Такой процесс соответствует определению взрыва, согласно которому, произведение продолжительности реакции (tр) на скорость звука (${v}$) в реакторе меньше, чем линейный размер (L) очага горения: ${v}$tp < L. Взрыв легко ингибируется на любой стадии в силу цепного механизма реакции. Поэтому он был назван нами цепно-тепловым взрывом (ЦТВ).
На рис. 7 иллюстрируется управление взрывом смеси 30% Н2 с воздухом при 1 атм в коническом реакторе объемом 180 л, обтянутом снизу резиновой оболочкой [27, 40]. Представлена зависимость максимального давления при взрыве и времени достижения взрывной волной вершины конуса от начального содержания ингибитора (пропилена). Инициатором служил взрыв 3 г гексогена на кронштейне в нижней части оболочки. Показания датчиков, расположенных вдоль образующей конуса, регистрировались на осциллографе, соединенном с ЭВМ. При инициировании взрыва в отсутствие ингибитора давление на вершине конуса ровно 1000 бар. Видно, что при увеличении содержания ингибитора в исходной смеси величина скачка давления падает. В области 0.95–1% ингибитора падение максимального давления резко усиливается, и при наличии более 2% пропилена взрыв газа не регистрируется. Видно также, что падение максимального давления взрыва сопровождается увеличением времени достижении взрывной волной вершины конуса. Ингибированием предотвращаются также взрыв метана (например, в испытательном штреке) [27].
Рис. 7.
Подавление инициированного взрыва водородо-воздушной смеси ингибитором; 1 – давление на вершине конуса, 2 – время достижения ударной волной вершины конуса, 3 – результаты повторных измерений спустя 60 дней.

Обратим внимание на то, что высокая эффективность ингибирования определяется не только большой скоростью реакционной способностью олефинов, но также тем, что скорость акта ингибирования находится в показателе степени экспоненты выражения (17) скорости цепной реакции. В реакциях с легко разветвляющимися цепями (горение силана, треххлористого азота) взрыв регистрируется даже при давлениях в области сотых долей атмосферного давления.
8. ОПРЕДЕЛЯЮЩАЯ РОЛЬ РЕАКЦИОННЫХ ЦЕПЕЙ В ДЕТОНАЦИИ
Детонация проявляется в совместном продвижении по горючей смеси поддерживающих друг друга взрывной реакции и ударной волны со сверхзвуковой постоянной скоростью. По чрезвычайно большим скоростям реакций, по газодинамике, по роли в технике и проявлениям детонация качественно отличается от других режимов горения. Участие реакционных цепей в детонации допускалось в ранних работах [41, 42], авторы которых, однако, позднее отрицали роль цепей в условиях значительного саморазогрева [2–4] и, значит, в условиях детонации. Наличие активных частиц в детонации отмечено авторами [43–46], считавшими источником тепловыделения тримолекулярную рекомбинацию НЦ в конце реакции. Однако, при этом развитие процесса гипотетически представлялось моделью одностадийной реакции валентно-насыщенных молекул. В работах этих авторов, как и в других работах, выполненных в соответствии прежней теорией детонации, учитывались реакции только исходных молекул (например, [22, 43–52]), которые, как было показано выше, не способны без участия свободных атомов и радикалов обеспечить режим горения. Температурную зависимость скорости реакции выражали аррениусовской функцией, которая как было показано выше, относится только к константе скорости, а экспоненциальная функция в формуле Аррениуса не описывает наблюдаемый сильный рост скорости при росте температуры. Кроме того, в горении и в детонации большая роль саморазогрева определяется тем, что он происходит не в конце горения, как это указано в [44–46], а на стадиях развития горения, благодаря чему и реализуется резкое самоускорение по закону (17). Величины скоростей ускорений реакций и причины больших величин в детонации ранее не рассматривались.
До наших работ [53–57] по выявлению и изучению цепного характера реакций в детонации, возможность ингибирования детонации отрицалась [47–52], что равносильно отрицанию ее цепной природы. Отрицание оказалось частично результатом неудачных попыток этих и других авторов ингибировать детонацию. Основной же причиной была приверженность теории нецепного горения. Даже после наших работ [53–58], в которых было показано предотвращение перехода горения в детонацию, разрушение стационарной детонационной волны и регулирование ее скорости путем ингибирования, встречаются публикации так называемого моделирования, в которых химический процесс представляют одностадийной реакцией с бессмысленными входными параметрами [16, 59].
На нецепной характер реакции в детонации, казалось бы, указывало согласие измеренных скоростей детонации водородо-воздушных смесей с рассчитанными в допущении о реакции в одну стадию, в том числе, по известной формуле:
где γ – отношение теплоемкостей смеси при постоянном объеме и постоянном давлении, Q – теплота реакции на единицу массы смеси [52]. Эта формула, однако, противоречит результатам наших исследований, в том числе данным по ингибированию детонации: разрушению стационарной детонационной волны и регулирования ее скорости с помощью малых примесей углеводородов [53–58], вступающих в реакции аналогичные(V) и (Vа). Согласие же формулы (20) с экспериментом определяется большой скоростью цепной реакции в отсутствие ингибитора, достигаемой благодаря высоким концентрациям активных частиц и большим скоростям их реакций.Предотвращение путем ингибирования перехода горения в детонацию и варьирование преддетонационного расстояния проводились в трубе диаметром 2.6 см [27, 57]. Наряду с искровым инициированием для турбулизации использовали спираль Щелкина. В модели прямоточного воздушно-реактивного двигателя турбулизация смеси водорода с нагретым воздухом и с ингибитором происходила при прохождении струи через смесители [27, 54, 55, 57]. Измеренные скорости и концентрационные пределы детонации в отсутствие ингибитора находятся в согласии с литературными данными. Из рис. 8 видно, что по мере увеличения содержания ингибитора концентрационные пределы перехода горения в детонацию сужаются, и при присадках более 2.6% (об.) переход предотвращается при любых соотношениях концентраций водорода и воздуха. При содержаниях ингибитора выше 8.5% подавляется также дефлаграционное горение. Обрыв цепей протекает в результате замены атомов Н на малоактивные радикалы по реакциям типа (VIII). В трубке меньшего диаметра с помощью ингибитора варьировали время задержки детонации.
Рис. 8.
Зависимости концентрационных пределов перехода горения водородо-воздушной смеси от содержания ингибитора в детонацию рабочей модели прямоточного воздушно-реактивного двигателя; 1 – горение без перехода в детонацию, 2 – детонация.

Была выявлена также определяющая роль цепной лавины в стационарной детонации [27, 56–58]. Реактор – секционная стальная труба длиной 15 м и диаметром 10.1 см – соединялся с узкой секцией, в которой с помощью искры создавалась инициирующая детонация стехиометрической смеси Н2 с О2. Отсюда детонационная волна входила в реактор с исследуемой смесью. Фронт пламени, ударная волна и детонация регистрировались с использованием расположенных вдоль трубы 8 пьезоэлектрических датчиков, и напротив них фотодиодов. Система датчиков, соединенных с пятью четырехканальными осциллографами, позволяла регистрировать раздельно детонацию, фронт пламени и ударную волну, скорости которых определяли по осциллограммам. Из рис. 9 видно, что детонационная волна смеси 2Н2 с О2 после вхождения в реактор с водородо-воздушной смесью в отсутствие ингибитора и прохождения расстояния 0.9–1.0 м продвигается с постоянной скоростью (прямая 1). Из наложения точек, обозначенных крестиками и кружками, друг на друга на прямой 1 (рис. 9) видно, что сигналы давления и свечения в каждом сечении вдоль всей трубы регистрируются одновременно, что присуще детонации. Скорость волны (1985 ± 5 м/с) находится в хорошем согласии с известными экспериментальными данными [4, 50, 52]. При прерывании цепной лавины с помощью 3% пропилена волна детонации замедляется, и на расстоянии 6 м фронт горения начинает отставать от ударной волны. Детонация распадается на затухающую ударную волну (кривая 4) и все больше отстающую от нее волну горения (кривая 5).
Рис. 9.
x–t – Диаграммы в смеси 30% Н2 с воздухом; 1, 2 и 3 – волны детонации без примесей, в присутствии 3% СО2 и СF4 соответственно, 4 – ударная волна, 5 – волна взрывной реакции.
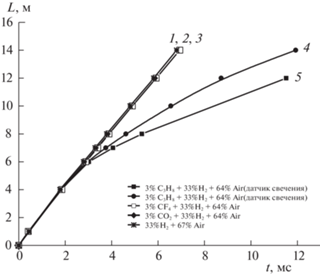
Вызванное ингибированием уменьшение интенсивности хемилюминесценции и скачка давления взрыва по мере продвижения волны вдоль реактора показывают также осциллограммы. На них виден также распад детонации на волну горения и ударную волну.
9. СКОРОСТИ, САМОУСКОРЕНИЯ РЕАКЦИИ И КОНЦЕНТРАЦИИ АКТИВНЫХ ЧАСТИЦ В ДЕТОНАЦИИ
Скорость детонации смеси 30% Н2 с воздухом составляла ~2 × 105 см [54, 56–58]. Ширина зоны пламени, согласно [22, 52] и нашим оценкам, не больше 0.1 см. Соответственно, продолжительность нахождения смеси в зоне пламени <10–6 с. Адиабатическая температура равна 2390 К. При допущении 10% тепловых потерь температура пламени равна 2180 К. В этих условиях концентрация О2 равна 5 × 1018 молек/см3. Скорость, полученная делением этой концентрации на время 10–6 с, равна 5 × 1024 молек/(см3 с), что в 7 × 108 раз больше скорости при 843 К. Таким образом, меньше, чем за миллионную долю секунды реакция ускоряется в сотни миллионов раз. Константа же скорости лимитирующей стадии k1 увеличивается при таком повышении температуры лишь в 450 раз. Из этих результатов также следует, что ускорение реакции горения вызвано фактически только ростом концентрации атомов Н, в соответствии с законом экспоненты с положительным показателем степени (фактор Больцмана), т.е. с уравнением (16). Поделив величину скорости реакции на произведение концентрации О2 (4.9 × 1018 молек/см3) и k1= 6.5 × 10–12 молек/(см3 с), получаем концентрацию атомарного водорода, равную 1.35 × 1017 атом /см3. Это значит, что в детонации ~7% исходного водорода превращены в атомы Н благодаря цепному механизму.
В свете описанных выше результатов можно определить детонацию, как режим горения, в котором химический процесс представляет собой цепную реакцию, скорость которой зависит от времени и от температуры по закону экспоненты с положительным показателем степени (фактор Больцмана с энергией активации лимитирующей стадии). В детонации водорода с воздухом характеристическое время реакции меньше микросекунды, благодаря концентрациям атомов водорода, составляющим десятки процентов от текущих концентраций исходных реагентов. Такой механизм горения определяет продвижение в горючей смеси с постоянной сверхзвуковой скоростью поддерживающиеся друг друга волны взрывного горения и ударной волны.
Таким образом, получен ответ на кардинальный вопрос о причинах экстремальных скоростей и ускорений процессов взрыва и детонации. Химический процесс в детонации – это цепная реакция с характеристическим временем, меньшем микросекунды и с концентрацией НЦ, ненамного ниже текущих концентраций исходных реагентов. Такие концентрации НЦ создаются по цепному механизму за счет основной части энтальпии процесса. Экстремальные скорости и ускорения реакций определяются высокими концентрациями этих частиц и зависимостью от температуры в соответствии с законом экспоненты с положительным показателем степени (фактор Больцмана). Так определяются и скорости тепловыделения.
Выявление цепного характера процессов горения, взрыва и детонации, законов их протекания, позволяет управлять ими с помощью методов ингибирования и промотирования [25, 27, 57, 58].
После выявления цепной природы горения, взрыва и детонации публикуется много работ по численному моделированию цепного горения, относящихся обычно к горению водорода. В них за редчайшими исключениями ставится цель лишь описать ту или иную частную закономерность. Роли цепной лавины и раздельно саморазогрева не рассматриваются. В некоторых работах вопреки экспериментам, утверждается, будто воспламенение при атмосферном давлении вызвано только саморазогревом (например, [20]). Моделирование информативно, если направлено не на описание закономерности, а на выяснение ее причин или деталей (см., например, [60, 61]).
10. ВЛИЯНИЕ ГЕТЕРОГЕННЫХ РЕАКЦИЙ В ГОРЕНИИ И ПРОБЛЕМЫ МОДЕЛИРОВАНИЯ
Важной особенностью горения газов, определяемой цепным характером реакции, является также сильная зависимость распространения пламени от химических свойств контактирующей поверхности. Тепловая теория горения, не учитывающая цепной характер горения газов, роль поверхности реактора, сводила только к теплоотводу. В действительности же гетерогенная рекомбинация атомов и радикалов, сопровождающаяся обрывом реакционных цепей, тормозит процесс горения и соответственно тормозит распространение пламени. В работах [27, 62] зависимость скорости распространения пламени от химических свойств поверхности была обнаружена путем предварительного споласкивания реакционных трубок растворами разных кислот. В свою очередь пламя влияет на химические свойства поверхности и на скорости гетерогенных реакций, что проявляется также в изменении интенсивности горения одной и той же смеси в повторных опытах [2–4, 6, 27]. Гетерогенный обрыв цепей определяют также градиенты концентраций активных частиц и температуры. Поскольку концентрации НЦ и температура максимальны в окрестности оси пламени, то моделирование без учета градиентов сильно занижает рассчитанную скорость процесса. На примере окисления водорода показано, что моделирование без учета градиентов не описывает также переход горения во взрыв [63]. Достоверность результатов численного моделирования горения возможна только при учете гетерогенных процессов. Поскольку гетерогенные реакции пока не учитываются, то встречающаяся часто запись гамильтониана при “моделировании” горения не соответствует реальному учету градиентов. Между тем, чрезвычайно большие скорости реакций в горении осуществляются при больших градиентах температуры и концентраций НЦ. Поэтому учет градиентов необходим при описании взрыва и детонации, а также при описании перехода горения в эти режимы.
Стабильное горение достигается, например, при очень больших коэффициентах гетерогенной рекомбинации, при которых обрыв цепей лимитируется диффузией активных частиц к поверхности. В этом случае для моделирования следует сначала доказать, что обрыв, действительно, лимитируется скоростью диффузии, и затем рассчитать эту скорость диффузии. Из полученных данных следует, что регулируемое изменение химических свойств поверхности можно использовать как метод управления горением.
Обобщены результаты комплексных исследований, в которых установлено, что в явлениях распространения пламени, взрыва и детонации газов химические процессы, вопреки прежним представлениям, являются цепными реакциями. Обсуждены кинетические законы неизотермических цепных процессов, решающая роль изменения концентраций свободных атомов и радикалов в чрезвычайно сильной температурной зависимости скорости и ускорении реакции. Показано, что благодаря специфике температурной зависимости скорости реакции и высоким концентрациям атомов и радикалов, характеристическое время процесса в ламинарном пламени меньше десятитысячной доли секунды, в детонации оно меньше микросекунды. Выяснены особенности перехода горения из одного режима в другой. Разработаны эффективные химические методы управления всеми режимами горения.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования РФ (соглашение № 075-15-2020-806 от 29 сентября 2020 г.) и за счет субсидий, выделенных ИСМАН на выполнение госзадания FFSZ-2022-0012 (рег. № 1021071612845-2-1.4.3; 2.5.1).
Список литературы
Семенов Н.Н. // Успехи физ. наук. 1940. Т. 23. № 1. С. 251.
Семенов Н.Н. Развитие теории цепных реакций и теплового воспламенения. М.: Знание, 1969. 95 с.
Семенов Н.Н. Избранные труды. М.: Наука, 2005. Т. 3. 499 с.
Lewis B., Von Elbe G. Combustion, Explosions and Flame in Gases. N.Y.-L.: Acad. Press, 1987. 592 p.
Baulch D.L., Bowman C.T., Cobos C.J. et al. // J. Phys. Chem. Ref. Data. 2005. V. 34. № 3. P. 757.
Srinivasan N.K., Michael J.V., Harding L.B., Klipperstain S.J. // Combust. Flame. 2007. V. 149. № 1/2. P. 104.
Зельдович Я.Б., Баренблат Г.И., Либрович В.Б., Махвиладзе Г.М. Математическая теория горения. М.: Наука, 1980. 478 с.
Химическая энциклопедия. М.: Сов. Энциклопедия, 1988. Т. 1. Статья “Горение”, с. 595.
Физическая энциклопедия. М.; Сов. Энциклопедия, 1988, Т. 1. Статья “Горение”, с. 515.
Большой энциклопедический словарь “Физика”. М.: Большая Российская энциклопедия, 1998. Статья “Горение”, с. 134.
Гельфанд Б.Е. // Физика горения и взрыва. 2002. Т. 38. № 5. С. 101.
Франк-Каменецкий Д.А. Основы макрокинетики, диффузия, теплопередача в химической кинетике. Долгопрудный: Интеллект, 2008. 407 с.
Кукин П.П., Юшин В.В., Емельянов С.Г. // Теория горения и взрыва. М.: Изд-во “Юрайт”, 2012. 435 с.
Палесский Ф.С., Фурсенко Р.В., Минаев С.С. // Физика горения и взрыва. 2014. Т. 50. № 6. С. 3.
Frassoldaty A., Couci A., Furavelly T. // Comb. Flame. 2010. V. 157. P. 2.
Frolov S.M., Shamshin I.V., Dubrovsrii F.V., Medvedev S.N. // Proceedings of International Conference on Transient Combustion and Detonation. Moscow: TORUS PRESS, 2014. P. 204.
Kogan I., Sivashinsky G. // Ibid. P. 163.
Сабденов К.И. // Хим. физика. 2017. Т. 36. № 11. С. 39.
Бабкин В.С., Сеначин П.К. Процессы горения газов в ограниченных объемах. Барнаул: Изд-во Алт.ГТУ, 2017. 143 с.
Варнатц Ю., Маас У., Дибба Р. Горение. Физические и химические аспекты, моделирование, эксперименты, образование загрязняющих веществ. М.: Физмат лит., 2003. 351 с.
Kim N.J. // Comb. Flame. 2009. V. 156. № 1. P. 132.
Chung K. Law Combustion Physics. Cambridge, New York: Cambridge University Press, 2006. 722 p.
Азатян В.В. // Успехи химии. 1999. Т. 62. № 12. С. 1122.
Азатян В.В., Вагнер Г.Г., Ведешкин Г.К. // Журн. физ. химии. 2004. Т. 78. № 6. С. 1036.
Азатян В.В., Павлов В.А., Шаталов О.П. // Кинетика и катализ. 2005. Т. 46. № 6. С. 835.
Азатян В.В. // Журн. физ. химии. 2011. Т. 85. № 8. С. 1405.
Азатян В.В. // Цепные реакции горения, взрыва и детонации в газах. М.: Изд-во РАН, 2020. 360 с.
Петрова Л.Д., Азатян В.В., Баратов А.Н. и др. Сб. Горение и взрыв. 1977. М.: Наука, 2020. С. 625.
Вильямс Ф.А. Теория горения. М.: Наука, 1971, 615 с.
Кондратьев В.Н., Никитин Е.Е. Химические процессы в газах. М.: Наука, 1981, 262 с.
Мержанов А.Г., Хайкин Б.И. Теория волн горения в гомогенных средах. Черноголовка: Изд-во Института структурной макрокинетики РАН, 1992. 160 с.
Азатян В.В. // Кинетика и катализ. 2015. Т. 56. № 1. С. 3.
Азатян В.В. // Там же. 1977. Т. 18. № 2. С. 282.
Азатян В.В., Шавард А.А. // Изв. АН СССР. Сер. хим. 1977. Т. 42. № 11. С. 2460.
Азатян В.В. // Кинетика и катализ. 2020. Т. 61. № 3. С. 291.
Гершензон Ю.М., Глебова О.Н., Азатян В.В. // Докл. АН СССР. 1966. Т. 168. № 4. С. 851.
Korobeinichev O.P., Shvartsberg V.M., Shmakov A.G. et al. // Proc. Comb. Institute. 2007. V. 31. P. 2741
Азатян В.В., Калканов В.А., Мержанов А.Г., Шавард А.А. // Хим. физика. 1987. Т. 6. № 12. С. 1696.
Азатян В.В., Вартанян А.А., Калканов В.А. Шавард А.А. // Там же. 1989. Т. 8. № 11. С. 1290.
Азатян В.В., Набоко И.М., Петухов В. А. и др. // Докл. РАН. 2004. Т. 394. № 1. С. 61
Lewis B. // J. Amer. Chem. Soc. 1930. V. 52. P. 3120, 3915.
Семенов Н.Н. Цепные реакции. Л.: Госхимтехиздат, 1934. 555 с.
Зельдович Я.Б. //ЖЭТФ. 1940. Т. 10. № 2. С. 524.
Зельдович Я.Б. Избранные труды. М.: Наука, 1984. 374 с.
Von Neumann J. // Progress report on the theory of detonation waves. 1943, OSRD Rept. № 549.
Doring W. // Ann. Physik. 1943. V. 43. P. 421.
Соколик А.С. Самовоспламенение, пламя и детонация в газах. М.: Изд-во АН СССР, 1960. 428 с.
Progress in Astronautics and Aeronautics. Advances in Combustion Science: in Honor of Zeldovich, 1997. V. 173. P. 95.
Гельфанд Б.Е. // Физика горения и взрыва. 2002, Т. 38. № 5. С. 101.
Нетлетон М. Детонация в газах. М.: Мир, 1989. 278 с.
Митрофанов В.В. Детонация гомогенных и гетерогенных систем. Новосибирск: Изд-во Сибирского отделения РАН, 2003. 199 с.
Зельдович Я.Б., Компанеец А.С. Теория детонации. М.: Гостехиздат, 1958. 268 с.
Азатян В.В., Калканов В.А., Романенко Н.Т., Шавард А.А., Федосов Л.Н. Пат. РФ № 2028369, 1995.
Azatyan V.V., Wagner G.G., Vedeshkin G.K. Gaseous and Heterogeneous Detonations. M.: ENAS Publishers, 1999. P. 331.
Azatyan V.V., Ayvazyan R.G., Vedeshkin G.K., Iskra V.A. // Proc. Intern. Symp. “Hydrogen 12”. Buenos Aires, 1998. P. 15.
Азатян В.В., Бакланов Д.И., Гвоздева Л.Г. и др. // Докл. РАН. 2001. Т. 376. № 1. С. 55.
Азатян В.В., Вагнер Г.Г., Ведешкин Г.К. // Журн. физ. химии. 2004. Т. 78. № 6. С. 1036.
Азатян В.В., Абрамов С.К., Прокопенко В.М. и др. // Кинетика и катализ. 2013. Т. 54. № 5. С. 553.
Дубровский А.В., Иванов В.С., Зангиев А.Э., Фролов С.М. // Хим. физика. 2016. Т. 35. № 6. С. 49.
Киверин А.Д., Яковенко И.С., Фортов В.Е. // Докл. РАН. 2018. Т. 489. № 5. С. 461.
Киверин А.Д., Смыгалина А.Е., Яковенко И.С. // Хим. физика. 2020. Т. 39. № 8. С. 9.
Азатян В.В., Прокопенко В.М., Абрамов С.К. // Журн. физ. химии. 2019. Т. 93. № 4. С. 622.
Андрианова З.С., Иванова А.Н., Азатян В.В. // Кинетика и катализ. 2002. Т. 43. № 2. С. 182.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии