

Журнал физической химии, 2022, T. 96, № 10, стр. 1490-1498
Исследование процесса атомно-слоевого осаждения оксида молибдена и титан-молибденовых оксидных пленок методом кварцевого пьезоэлектрического микровзвешивания
А. М. Максумова a, И. М. Абдулагатов a, *, Д. К. Палчаев a, М. Х. Рабаданов a, А. И. Абдулагатов a
a Дагестанский государственный университет
367000 Махачкала, Россия
* E-mail: ilmutdina@gmail.com
Поступила в редакцию 19.01.2022
После доработки 19.04.2022
Принята к публикации 20.04.2022
- EDN: LNDUAG
- DOI: 10.31857/S0044453722100181
Аннотация
Проведено исследование процесса термического атомно-слоевого осаждения (АСО) пленок оксида молибдена (MoOx) с использованием MoOCl4 и H2O, а также титан-молибденовых оксидных (TixMoyOz) тонких пленок с использованием TiCl4, MoOCl4 и H2O. Процесс роста пленки исследовали in situ методом кварцевого пьезоэлектрического микровзвешивания (КПМ) в диапазоне температур от 115 до 180°С. Рассмотрены процессы АСО пленок TixMoyOz с различным соотношением субциклов TiCl4–H2O и MoOCl4–H2O в суперцикле. Во всех случаях установлен линейный рост пленки с количеством АСО циклов. Показано, что поверхностные реакции галогенидов и H2O носили самоограничивающийся характер. Согласно данным КПМ, сделан вывод о возможности использования рассмотренной химии поверхности для осаждения тонких пленок MoOx и TixMoyOz. Установлены потенциальные области применения данных тонких пленок: катализ, электрохромные устройства, литий-ионные аккумуляторы, антибактериальные покрытия и т.д.
Известно, что диоксид титана (ТiО2) широко распространен в природе, не токсичен и обладает фотокаталитическими свойствами. Значение ширины запрещенной зоны TiO2 в зависимости от кристаллической структуры находится в пределах от 3.0 до 3.4 эВ. Это ограничивает его область активации ультрафиолетовой областью, которая составляет лишь ≈3% солнечного спектра [1]. Поэтому разработка активного в видимой области света диоксида титана – одна из ключевых задач в области фотокатализа полупроводников [2]. Одним из подходов к модификации оптических свойств TiO2 служит легирование ионами переходных металлов [3]. Легированные покрытия можно получить различными газофазными методами, такими как химическое осаждение из газовой фазы (ХОГФ) [4], магнетронное распыление [5], испарение лазерным лучом [6] и т.д. Метод атомно-слоевого осаждения (АСО) под названием “молекулярное наслаивание” был впервые разработан в 60-е годы прошлого столетия советскими учеными В.Б. Алесковским и С.И. Кольцовым [7]. Данная технология позволяет на атомарном уровне контролировать толщину и состав получаемых пленок [8, 9]. Поэтому АСО нашло широкое применение для получения и в том числе легированных тонких пленок. В прошлом для легирования АСО TiO2 были успешно использованы ванадий [10], углерод [11], азот [12–14], ниобий [15], сера [16], цинк [17], фтор [18], тантал [19] и т.д. В данной работе впервые исследовали процесс термического АСО оксида титана, легированного молибденом, с использованием тетрахлорида титана, окситетрахлорида молибдена и воды. Возможность использования оксидных сплавов титана и молибдена в фотокатализе ранее была продемонстрирована в работах [20, 21]. Кроме этого, пленки TixMoyOz могут найти применение в литий-ионных аккумуляторах [22], газовых сенсорах [23] и т.д.
Процесс АСО TixMoyOz, рассмотренный в данной работе, является комбинацией двух процессов: АСО TiO2 и MoO3. В настоящее время АСО TiO2 продемонстрировано с использованием множества различных типов прекурсоров. Ниже приведены только некоторые из них: тетрахлорид титана (TiCl4) [24]; изопропоксид титана (Ti(OiPr)4) [25]; тетракисдиметиламинотитан (Ti(NMe2)4) [26]; бис-(изопропоксид)-бис-(2,2,6,6-тетраметилгептан-3,5-дионат) титана (Ti(OiPr)2(thd)2) [27] и др. в комбинации с такими окислителями как H2O, О3, H2O2 и т.д. [28]. В качестве прекурсоров для получения пленок MoOx ранее были использованы гексакарбонил молибдена (Mo(CO)6) [29]; бис-этилбензол молибдена (MoC16H20) [30]; диоксо-бис-(N,N'-диизопропилацетоамидинат) молибдена (VI) (MoO2(iPr2amd)2) [31]; бис-(трет-бутилимидо)-бис-(диметиламино)молибден (VI) (Mo(NtBu)2(NMe2)2) [32]; окситетрахлорид молибдена (VI) (MoOCl4) [33]; диоксо-бис-(2,2,6,6-тетраметилгептан-3,5-дионато)молибден (VI) (MoO2(thd)2) [34]; диоксо-бис-(N,N'-трет-бутилацетоамидинато)молибден (VI) MoO2(tBuamd)2 [35]; Mo(CpSiMe3)(CO)2(2-метилаллил) [36], в комбинации с H2O, O3 и H2O + O3. Тонкие пленки MoOx находят применение в катализе [37, 38], электрохромных устройствах [39], литий-ионных батареях [40], газовых сенсорах [41], в качестве антибактериальных покрытий [42].
В сравнении с часто используемыми в АСО TiO2 и МоОх органометаллическими прекурсорами, их галогениды обладают достаточным давлением паров при комнатной температуре или могут быть относительно легко переведены в газовую фазу нагревом. В связи с этим в данной работе АСО TixMoyOz проводили с использованием TiCl4, MoOCl4 и H2O. В качестве альтернативного прекурсора молибдена (VI) может быть использован более термически стабильный диоксидихлорид молибдена МоО2Cl2 [43, 44], однако он обладает более высокой температурой плавления (175°С) [45].
Цель данной работы – исследование поверхностных процессов, происходящих при АСО оксида молибдена и титан-молибденовых оксидных тонких пленок при помощи КПМ в диапазоне температур 115–180°С.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
АСО оксида молибдена и титан-молибденовых оксидных пленок проводили на оборудовании компании ООО “АСО НаноТех”. Упрощенная схема экспериментальной установки приведена на рис. 1.

Для АСО использовали вакуумную камеру с горячими стенками, выполненную из стали марки AISI 304L, которую продували потоком газа- носителя. В качестве газа-носителя использовали азот особой степени чистоты от ООО “Гермес-газ” (N2, 99.999%). Напуск паров прекурсоров осуществляли посредством открытия пневматических дозирующих клапанов, которые установлены между контейнером с прекурсором и реактором. Последовательность и продолжительность напуска прекурсоров задавали программой. Парциальные давления прекурсоров регистрировали встроенным на выходе из реактора датчиком давления как показано на схеме. С помощью регулятора расхода газа (РРГ) давление в реакторе поддерживали азотом на уровне ~1.0 Торр. При этом РРГ устанавливали на расход в 100 см3/мин. TiCl4 и MoOCl4 перед экспериментом загружали в контейнеры для дозирования в атмосфере аргона. Чистота TiCl4 (Sigma-Aldrich, кат. номер 7550450) и MoOCl4 (Sigma-Aldrich, кат. номер 13814750) составляла ≥99.0 и 97.0%, соответственно. Воду использовали хроматографического класса чистоты (Fisher Chemical, кат. номер W5-1). АСО проводили при температурах 115, 150 и 180°С. Во время АСО MoOCl4 грели до 60°С для сублимации и достижения достаточного давления паров. Температура плавления MoOCl4 составляет 105°С [45]. Известно, что MoOCl4 термически нестабилен и очень медленно [43] подвергается разложению при 25°С. Визуально изменение цвета прекурсора после нагревания в контейнере до 60°С не наблюдалось.
Изучение и оптимизацию процесса роста пленок проводили в АСО-установке, снабженной кварцевыми микровесами, позволяющими проводить исследование роста пленки в режиме реального времени (in situ). КПМ-оборудование, использованное в данной работе, схоже с описанным в работе [46]. КПМ-измерения выполняли с использованием электронного модуля STM-2 (Inficon). Корпус КПМ выполнен компанией Inficon. Разрешение кварцевых микровесов по массе составляет ~0.3 нг/см2. До начала осаждения пленок MoOx или TixMoyOz кристалл КПМ покрывали пленкой АСО Al2O3, для этого использовали триметилалюминий (ТМА, Al(CH3)3) и воду. ТМА с чистотой 97% (Sigma-Aldrich, кат. номер 257222) использовали при комнатной температуре без нагрева. В процессе роста Al2O3 наблюдали линейный прирост массы с количеством циклов со средним приростом массы за цикл ~33.0 нг/см2, что хорошо согласуется с ранее опубликованными данными [47]. Значения погрешностей, полученные для каждой точки на кривых насыщения, представляют разброс данных по 10 точкам для разных экспериментов. Относительная погрешность для данных измерений составляет 1.07%. Активная площадь кварцевого кристалла, на которой происходило осаждение, составляла 0.65 см2 и по прилагаемой программе пересчитывалась на площадь 1 см2.
Время напуска и продувки прекурсоров во время одного АСО цикла MoOx обозначали как t1/t2/t3/t4, где t1 – время напуска паров MoOCl4; t2 и t4 – время продувки; t3 – время напуска паров H2O. Один АСО суперцикл осаждения TixMoyOz обозначали как t1/t2/t3/t4/t5/t6/t7/t8, где t1 – время напуска паров TiCl4; t2, t4, t6, t8 – время продувки; t3, t7 – время напуска паров H2O; t5 – время напуска паров MoOCl4. Соотношение субциклов TiCl4–H2O и MoOCl4–H2O в процессе АСО TixMoyOz варьировали, меняя количество t5/t6/t7/t8 субциклов в суперцикле. Парциальные давления MoOCl4, TiCl4 и H2O при времени напуска в течение 1 с составляли ~5, ~15 и ~50 мТорр, соответственно. Термохимические расчеты проводили с использованием программы HSC Chemistry.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Атомно-слоевое осаждение МоОх
Рост пленки MoOx осуществлялся за счет поверхностных реакций паров MoOCl4 и H2O. Термохимические расчеты для газофазной реакции MoOCl4(г) + 2H2O(г) → MoO3 + 4HCl(г) при 150°C (∆G = –17.23 ккал/моль) показывают, что данная реакция может протекать самопроизвольно. Процесс АСО MoOx схематично можно представить в виде циклически повторяющихся поверхностных полуреакций:
(A)
$\begin{gathered} ](--{\text{OH}})_{x}^{*} + {\text{MoOC}}{{{\text{l}}}_{4}}({\text{г}}) \to \\ \to \;]{{(--{\text{O}})}_{х}}{\kern 1pt} --{\kern 1pt} {\text{MoO}}({\text{Cl}})_{{4 - x}}^{*} + х{\text{HCl}}({\text{г}}), \\ \end{gathered} $(Б)
$\begin{gathered} ]{{(--{\text{O}})}_{х}}{\kern 1pt} --{\kern 1pt} {\text{MoO}}({\text{Cl}})_{{4 - x}}^{*} + (4--х){{{\text{H}}}_{{\text{2}}}}{\text{O}}({\text{г}}) \to \\ \to \;]{{(--{\text{O}})}_{x}}{\kern 1pt} --{\kern 1pt} {\text{MoO}}({\text{OH}})_{{4 - x}}^{*} + (4--х){\text{HCl}}({\text{г}}), \\ \end{gathered} $На рис. 2 представлены КПМ-данные о зависимости прироста массы от времени осаждения в процессе АСО MoOx при 115, 150 и 180°С.

Процесс проводили с временными параметрами напуска и продувки 1/30/1/30. При температуре АСО 115°С прирост массы был наименьшим, максимальный прирост массы наблюдался при 180°С. Скорость прироста массы при 150 и 115°С была примерно одинаковой. При 150 и 180°С наблюдали линейный рост пленки с количеством АСО-циклов и высокую повторяемость поверхностных процессов от цикла к циклу. Повторяемость процесса и линейность роста пленки была хуже при 115°С.
Зависимость прироста массы, приходящегося на один цикл АСО MoOx, от продолжительности напуска MoOCl4 и H2O при температуре АСО 150°С приведена на рис. 3. Данные эксперименты были проведены для определения самонасыщаемости поверхностных полуреакций (А) и (Б). Результаты для MoOCl4 были получены c использованием временных параметров одного цикла α/30/1/30, где α – варьируемое время напуска паров MoOCl4, 1 с – фиксированное время напуска паров H2O и 30 с – время продувки прекурсоров. Результаты для H2O были получены c использованием временных параметров одного цикла 1/30/α/30, где α – варьируемое время напуска паров H2O, 1 с – фиксированное время напуска паров MoOCl4.
Рис. 3.
Зависимости прироста массы от длительности напуска паров MoOCl4 и H2O в процессе АСО MoOx при 150°С.

Из рис. 3 следует, что прирост массы за цикл достигает насыщения уже при времени напуска паров MoOCl4 ~1.0 с. Кривая насыщения для H2O имеет более плавный характер, возможно, связанный с задержкой воды в реакторе и необходимостью более длительной продувки после напуска воды.
На рис. 4 представлен наблюдаемый при 150°С приближенный вид сигнала КПМ в ходе АСО MoOx с параметрами цикла 1/30/1/30. При напуске MoOCl4 результирующий прирост массы после напуска MoOCl4 и шага продувки составил ~10 нг/см2. На стадии продувки происходит некоторое снижение массы, что свидетельствует о нестабильности поверхностных комплексов и/или о медленной десорбции продуктов реакции. Напуск H2O приводит к резкому снижению массы на 3.0 нг/см2, при этом результирующий прирост массы составляет 7.0 нг/см2 за цикл. Постоянную роста (δ, Å/цикл) можно получить из уравнения δ = ΔmАρ–1, где ΔmА – прирост массы за один цикл (7.0 нг/см2), ρ – плотность кристаллического МоО3 (4.69 г/см3); постоянная роста δ ~ 0.15 Å/цикл, что близко к значению 0.1 Å/цикл, полученному ранее для АСО MoOx при 300°С с использованием тех же прекурсоров [33]. Толщину одного монослоя MoO3 можно рассчитать из уравнения h = = (M/(NAρ))1/3, где M – молярная масса MoO3, NA – число Авагадро, и ρ – плотность кристаллического MoO3. Из приведенного уравнения толщина монослоя кристаллического MoO3 составляет 3.7 Å. Расчетным путем также определили ожидаемый прирост массы на КПМ для одного монослоя MoO3 (~174 нг/см2). Из этого следует, что осаждение пленки оксида молибдена в данном случае происходит в субмонослойном режиме. Прирост массы при напуске MoOCl4 и ее снижение при дозировании H2O соответствуют предложенной химии осаждения. Отношение общего прироста массы за один АСО цикл MoOCl4/H2O (∆mA) к приросту массы после напуска MoOCl4 (∆mB) составляет ${{R}_{{{\text{Mo}}{{{\text{O}}}_{{\text{3}}}}}}}$ = 7.0/10.0 = 0.7. Из уравнения: ${{R}_{{{\text{Mo}}{{{\text{O}}}_{{\text{3}}}}}}}$ = M(MoO3)/[M(MoOCl4) – xM(HCl)], где М – молярная масса, можно рассчитать долю прореагировавших –ОН-групп (x). Полученное значение x = 1.3 соответствует случаю, когда взаимодействие MoOCl4 происходит в основном с одной гидроксильной группой.
Рис. 4.
Изменение прироста массы (∆m) в процессе попеременного напуска паров MoOCl4 и H2O при 150°С для АСО MoOx.

В табл. 1 рассмотрены три случая, когда рост MoOx осуществляется с участием одной, двух или трех поверхностных гидроксильных групп. При этом идеальным является случай, где х = 2, при котором количество регенерированных гидроксильных групп после напуска H2О (реакция (Б)) равно количеству прореагировавших (реакция (А)). Однако экспериментально полученное значение x = 1.3, что указывает на высокую вероятность монодентатного присоединения MoOCl4 (х = 1), в этом случае количество прореагировавших в реакции (А) гидроксильных групп становится меньше полученных в результате реакции (Б). Как было показано ранее, поверхностные реакции VOCl3 [48] или TMA [49] с H2О могут быть поликонденсацией (дегидратацией) соседних металгидроксильных групп с возникновением мостиковых связей. Такой вариант для системы MoOCl4/H2O представлен на рис. 5.
Таблица 1.
Возможные механизмы роста MoOx с расчетными значениями R = ∆mA/∆mВ
| х | Реакции | R |
|---|---|---|
| 1 | ]–OH* + MoOCl4(г) → ]–O–MoOCl$_{3}^{*}$ + HCl(г) ]–O–MoOCl$_{3}^{*}$ + 3H2O(г) → ](–O)–MoO–(OH)$_{3}^{*}$ + 3HCl(г) |
0.71 |
| 2 | ]–(OH)$_{2}^{*}$ + MoOCl4(г) → ](–O)2–MoOCl$_{2}^{*}$ + 2HCl(г) ](–O)2–MoOCl$_{2}^{*}$ + 2H2O(г) → ](–O)2–MoO–(OH)$_{2}^{*}$ + 2HCl(г) |
0.80 |
| 3 | ]–(OH)$_{3}^{*}$ + MoOCl4(г) → ](–O)3–MoOCl* + 3HCl(г) ](–O)3–MoOCl* + H2O(г) → ](–O)3–MoO–OH* + HCl(г) |
0.87 |
Рис. 5.
Схема возможных поверхностных процессов взаимодействия паров MoOCl4 и H2O в процессе АСО МоOx при 150°С.

Для этого случая расчетное значение R = 0.7 и совпадает с экспериментальным. Все представленные расчеты предполагают рост MoOх без примесей хлора.
Вследствие обратимости поверхностных химических реакций осаждаемые оксиды могут быть переведены в газовую фазу в результате хемосорбции продукта реакции – HCl [50, 51]. Согласно термохимическим расчетам, процессы травления оксида молибдена в процессе реадсобции продукта реакции (HCl) или продукта разложения прекурсора MoOCl4 в контейнере (Cl2) маловероятны, так как энергии Гиббса для реакций MoO3 + 4HCl(г) → MoOCl4(г) + 2H2O(г) (ΔG(150°С) = 18.0 ккал) и MoO3 + 2Cl2(г) → MoOCl4(г) + O2(г) (ΔG (150°С) = 32.25 ккал) положительны. Это свидетельствует о том, что поверхностные реакции не обратимы, поэтому продукты реакции на стенках реактора не должны влиять на процессы, происходящие на КПМ. Также энергии Гиббса имели положительные значения и для случаев, когда возможным продуктом реакции был MoO2Cl2.
Приведенные выше экспериментальные данные указывают на то, что химия поверхности MoOCl4 и H2O может быть использована для осаждения сплава TixMoyOz.
Атомно-слоевое осаждение TixМоyОz
АСО TixMoyOz осуществляли за счет поверхностных реакций паров TiCl4, MoOCl4 и H2O в заданной последовательности. Полученные пленки обозначили как 1Ti1MoО, 1Ti7MoO и 2Ti7MoO, где коэффициенты соответствуют количеству субциклов TiCl4/H2O и MoOCl4/H2O в суперцикле. Для осаждения 1Ti1MoО использовали восьмиступенчатый АСО суперцикл, состоящий из последовательного напуска паров TiCl4, H2O, MoOCl4, H2O и продувок между ними (рис. 6).

Зависимость прироста массы, приходящегося на один суперцикл АСО TixMoyOz (1Ti1MoО), от продолжительности напуска паров TiCl4, MoOCl4, H2O при температуре АСО 150°С приведена на рис. 7.
Рис. 7.
Зависимость прироста массы (∆m) от длительности напуска паров TiCl4, MoOCl4 и H2O в процессе осаждения АСО TixMoyOz (1Ti1MoО) при 150°С.

Для определения самонасыщаемости реакции TiCl4 использовали временные параметры одного суперцикла α/30/2/30/1.5/30/2/30, где α – варьируемое время напуска паров TiCl4, 1.5 с – фиксированное время напуска паров MoOCl4, 2 c – время напуска паров H2O, 30 с – время продувки прекурсоров, а для самонасыщаемости реакции MoOCl4 – 1.5/30/2/30/α/30/2/30, где 1.5 с – время напуска паров TiCl4, α – варьируемое время напуска паров MoOCl4, 2 c – время напуска паров H2O, 30 с – время продувки прекурсоров. Из рис. 6 видно, что насыщение достигалось при времени напуска паров галогенидов ~1.0 с. На рис. 7 также представлены результаты по самонасыщаемости реакции H2O, полученные для времени напуска и продувки в суперцикле 1.5/30/α/30/1.5/30/α/30, где пары TiCl4 и MoOCl4 напускали в течение 1.5 с, α – варьируемое время напуска паров H2O, 30 с – время продувки прекурсоров. Из изложенного выше следует, что поверхностные реакции галогенидов имеют самоограничивающийся характер, а кривая насыщения для H2O имеет более плавный характер.
На рис. 8 приведены КПМ-данные по изменению массы при напуске и продувке паров реагентов в процессе роста пленки 1Ti1MoО при 150°С. Рост пленки осуществлялся с использованием временных параметров одного суперцикла 1/30/1/30/1/30/1/30. Прирост массы после TiCl4/H2O субцикла составил 17.0 нг/см2, что ниже значения прироста массы в процессе АСО TiO2 (20.2 нг/см2), полученного при схожих условиях роста. Прирост массы после MoOCl4/H2O-субцикла составил 20.0 нг/см2, что значительно выше полученного в процессе АСО MoOx (~7.0 нг/см2, рис. 3). Увеличение прироста массы за субцикл MoOCl4/H2O свидетельствует о повышении концентрации реакционноспособных поверхностных групп после TiCl4/H2O-субцикла.
Рис. 8.
Прирост массы (∆m) в процессе АСО титан-молибденовой оксидной пленки (1Ti1MoO) в зависимости от роста.
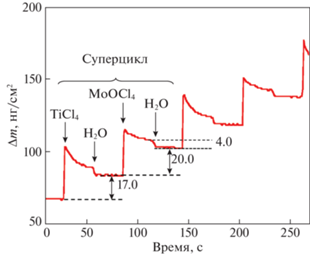
Из отношения общего прироста массы за субцикл (20.0 нг/см2) к приросту массы при напускe MoOCl4 (24.0 нг/см2) рассчитали количество гидроксильных групп (х), принявших участие в реакции поверхностного гидролиза (2.2), что указывет на бидентатное присоединение MoOCl4 к поверхностным функциональным группам в процессе роста 1Ti1MoO. Изменение механизма поверхностных реакций также может быть связано с формированием поверхностных донорно-акцепторных комплексов (–O)хTi ← :O = MoCl4, вследствие образования координационной связи между неподеленной электронной парой атома кислорода в молекуле MoOCl4 с вакантными d-орбиталями атомов титана [52].
Для увеличения относительного содержания молибдена в титан-молибденовой оксидной пленке АСО проводили с использованием одного субцикла TiCl4/H2O и семи субциклов MoOCl4/H2O в суперцикле (пленки 1Ti7MoО). На рис. 9 приведены данные КПМ, наблюдаемые в процессе АСО, проводимого с временными параметрами суперцикла 1/30/1/30/((1/30/1/30) × 7) при 150°С.
Рис. 9.
Изменение массы (∆m) для одного суперцикла в процессе АСО титан-молибденовой оксидной пленки (1Ti7MoO) с семью субциклами MoOCl4/H2O.

После субцикла TiCl4/H2O наблюдается общая потеря массы (~3.0 нг/см2). Примечательно, что потеря массы наблюдается как после напуска H2O, так и после TiCl4, что свидетельствует о процессах травления пленки. Прирост массы после семи субциклов MoOCl4/H2O составил 82.0 нг/см2. Общий прирост массы за один суперцикл составил ~79.0 нг/см2. С каждым последующим MoOCl4/H2O субциклом прирост массы постепенно снижается.
Потерю массы, наблюдаемую после напуска TiCl4, можно объяснить его взаимодействием с поверхностным оксидом молибдена и удалением Mo в виде оксихлоридов по схеме: MoO3 + + TiCl4(г) → TiO2 + MoOCl4(г), ΔG(150°C) = = ‒5.5 ккал/моль и/или 2MoO3 + TiCl4(г) → TiO2 + + 2MoO2Cl2(г), ΔG(150°C) = –6.4 ккал/моль (в перерасчете на один атом молибдена ΔG(150°C) = = –3.2 ккал/моль). Обе реакции термодинамически разрешимы при 150°С. Согласно справочным данным, температура сублимации MoO2Cl2 составляет 157°С [53], что близко к температуре осаждения. Сопутствующие реакции травления могут способствовать снижению содержания молибдена в осаждаемой пленке (отклонение от правила смесей), что наблюдалось ранее при осаждении других многокомпонентных АСО-пленок [10, 54]. Процессы “конвертирования” оксидов молибдена и титана могут быть схожи с процессами изоморфного замещения. Данные процессы можно объяснить [55] более низким значением стандартной энтальпии образования TiO2 (анатаз, –938.7 кДж моль–1) в сравнении с MoO3 (–744.6 кДж моль–1). Потеря массы после напуска H2O в субцикле TiO2 объясняется реакциями замещения Cl-лигандов поверхностных оксититанхлоридных и/или оксимолибденхлоридных групп. Формирование связей Мо–Cl происходит вследствие перехода части Cl-лигандов TiCl4 на неполностью удаленные с поверхности атомы молибдена. Возможно, что схожие процессы протекают также и при осаждении пленок 1Ti1MoO, где процесс “конвертирования” после напуска TiCl4 проявляется в меньшей степени.
На рис. 10 показан общий вид наблюдаемого при 150°С сигнала КПМ в процессе АСО TixMoyOz с разными соотношениями субциклов. При осаждении пленок пары прекурсоров металлов и воды напускались в течение 1 с и продувались в течение 30 с.
Рис. 10.
Прирост массы (∆m) в процессе АСО титан-молибденовых оксидных пленок с разным соотношением субциклов при 150°С.

На рис. 10 видна линейность роста пленок с количеством АСО-циклов, а также повторяемость процесса от цикла к циклу. Угол наклона линии прироста массы для 1Ti1MoО процесса выше, и, соответственно, скорость роста пленки в данном случае выше, чем для 1Ti7MoО и 2Ti7MoO. Скорость роста пленки увеличивается при увеличении количества TiCl4/H2O-субциклов (2Ti7MoO) и убывает с увеличением количества MoOCl4/H2O-субциклов (1Ti7MoO).
ЗАКЛЮЧЕНИЕ
In situ мониторинг процесса осаждения в диапазоне температур 115–180°С позволил установить линейность роста пленок MoOx и TixMoyOz с количеством циклов. Для обоих типов пленок поверхностные реакции галогенидов и H2O имели самоограничивающийся характер. Расчеты показали, что рост пленки MoOx происходит в субмонослойном режиме. В процессе реакции MoOCl4 с гидроксилированной поверхностью в основном наблюдается монодентатное присоединение, тогда как в сплаве в процессе АСО TixMoyOz (1Ti7MoO) эта реакция соответствовала бидентатному присоединению. В процессе роста пленок АСО TixMoyOz (1Ti7MoO) после субцикла TiCl4/H2O вместо ожидаемого прироста массы наблюдали ее снижение, которое объясняли “конвертированием” MoOx и TiO2 путем перехода хлор-лигандов TiCl4 на поверхностные оксимолибденовые группы и удалением Mo в газовую фазу в виде оксихлоридов. Для более детального изучения поверхностных процессов роста АСО MoOx и TixMoyOz запланировано проведение исследований полученных пленок с использованием инфракрасной и рентгеновской фотоэлектронной спектроскопии.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации (Государственное задание FZNZ-2020-0002).
Список литературы
Ren W., Ai Zh., Jia F. et al. // Appl.Catal. 2007. V. 69. № 3–4. P. 138. https://doi.org/10.1016/j.apcatb.2006.06.015
Fujishima A., Zhang X.T. // C. R. Chimie. 2006. V. 9. № 5–6. P. 750. https://doi.org/10.1016/j.crci.2005.02.055
Daghrir R., Drogui P., Robert D. // Ind. Eng. Chem. Res. 2013. V. 52. № 10. P. 3581. https://doi.org/10.1021/ie303468t
Dunnill C.W., Kafizas A., Parkin I.P. // Chem. Vap. Dep. 2012. V. 18. № 4–6. P. 89. https://doi.org/10.1002/cvde.201200048
Vahl A., Veziroglu S., Henkel B. et al. // Materials. 2019. V. 12. № 17. P. 2840. https://doi.org/10.3390/ma12172840
Al Mashary F.S., Felix J.F., Ferreira S.O. et al. // MSEB. 2020. V. 259. P. 114578. https://doi.org/10.1016/j.mseb.2020.114578
Малыгин А.А. // Сборник тезисов докладов III Международного семинара “Атомно-слоевое осаждение: Россия, 2021”, 2021. С. 13.
George S.M. // Chem. Rev. 2010. V. 110. № 1. P. 111. https://doi.org/10.1021/cr900056b
Puurunen R.L. // J. Appl. Phys. 2005. V. 97. № 12. P. 121301. https://doi.org/10.1063/1.1940727
Абдулагатов А.И., Максумова А.М., Палчаев Д.К. и др. // Журн. прикл. химии. 2021. Т. 94. № 7. С. 835. https://doi.org/10.1134/S1070427221070053
Xie Y., Zhao X., Chen Y. et al. // J. Solid State Chem. 2007. V. 180. № 12. P. 3576. https://doi.org/10.1016/j.jssc.2007.10.023
Tian L., Soum-Glaude A., Volpi F. et al. // J. Vac. Sci. Technol. A. 2015. V. 33. № 1. P. 01A141–1. https://doi.org/10.1116/1.4904025
Lee A., Libera J.A., Waldman R.Z. et al. // Adv. Sustainable Syst. 2017. V. 1. № 1–2. P. 1600041. https://doi.org/10.1002/adsu.201600041
Pore V., Heikkilä M., Ritala M. et al. // J. Photochem. Photobiol. 2006. V. 177. № 1. P. 68. https://doi.org/10.1016/j.jphotochem.2005.05.013
Niemela J.P., Yamauchi H., Karppinen M. // Thin Solid Films. 2014. V. 551. P. 19. https://doi.org/10.1016/j.tsf.2013.11.043
Pore V., Ritala M., Leskelä M. et al. // J. Mater. Chem. 2007. V. 17. № 14. P. 1361. https://doi.org/10.1039/B617307A
Su C.Y., Wang L.Ch., Liu W.S. et al. // Acs Appl. Mater. Interfaces. 2018. V. 10. № 39. P. 33287. https://doi.org/10.1021/acsami.8b12299
Pore V., Kivelä T., Ritala M. et al. // Dalton Trans. 2008. V. 45. P. 6467. https://doi.org/10.1039/B809953G
Choi J.H., Kwon S.H., Jeong Y.K. et al. // J. Electrochem. Soc. 2011. V. 158. № 6. P. B749. https://doi.org/10.1149/1.3582765
Huang J.-g., Guo X-t., Wang B. et al. // J. Spectros. 2015. V. 2015. P. 681850. https://doi.org/10.1155/2015/681850
Liu H., Lv T., Zhu Ch., Zhu Zh. // Sol. Energy Mater. Sol. Cells. 2016. V. 153. P. 1. https://doi.org/10.1016/j.solmat.2016.04.013
Zhang J., Huang T., Zhang L., Yu A. // J. Phys. Chem. C. 2014. V. 118. № 44. P. 25300. https://doi.org/10.1021/jp506401q
Galatsis K., Li Y.X., Wlodarski W. et al. // Sens. Actuators B Chem. 2002. V. 3. № 1–3. P. 276.
Кольцов С.И. // Журн. прикл. химии. 1969. Т. 42. № 5. С. 1023.
Dill P., Pachel F., Militzer C. et al. // J. Vac. Sci. Technol. A. 2021. V. 39. № 5. P. 052406. https://doi.org/10.1116/6.0001193
Kavan L., Tétreault N., Moehl Th., Graetzel M. // J. Phys. Chem. C. 2014. V. 118. № 30. P. 16408. https://doi.org/10.1021/jp030790+
Qi X., Jiang Yu., Detavernier C. et al. // J. Appl. Phys. 2007. V. 102. P. 083521. https://doi.org/10.1063/1.2798384
Niemela J.P., Marin G., Karppinen M. // Semicond. Sci. Technol. 2017. V. 32. № 9. P. 093005. https://doi.org/10.1088/1361-6641/aa78ce
Diskus M., Nilsen O., Fjellvå H. // J. Mater. Chem. 2011. V. 21. P. 705. https://doi.org/10.1039/C0JM01099E
Drake T.L., Stair P.C. // J. Vac. Sci. Technol. A. 2016. V. 34. https://doi.org/10.1116/1.4959532
Jurca T., Peters A.W., Mouat A.R. et al. // Dalton Trans. 2017. V. 46. P. 1172. https://doi.org/10.1039/C6DT03952A
Vos M.F.J., Bacco M., Thissen N.F.W. et al. // J. Vac. Sci. Technol. A. 2016. V. 34. P. 01A103. https://doi.org/10.1116/1.4930161
Kvalvik J.N., Jon B., Hansen P-A., Nilsen O. // J. Vac. Sci. Technol. A. 2020. V. 38. № 4. P. 042406. https://doi.org/10.1116/6.0000219
Mattinen M., King P.J., Khriachtchev L. et al. // Mater. Today Chem. 2018. V. 9. P. 17. https://doi.org/10.1016/j.mtchem.2018.04.005
Aidan R., Mouat A.R., Mane A.U. et al. // Chem. Mater. 2016. V. 28. № 6. P. 1907. https://doi.org/10.1021/acs.chemmater.6b00248
Nanayakkara C.E., Vega A., Liu G. et al. // Chem. Mater. 2016. V. 28. № 23. P. 8591. https://doi.org/10.1021/acsami.1c06204
Fransen T., Meer O., Mars P. et al. // J. Phys. Chem. 1976. V. 80. № 19. P. 2103. https://doi.org/10.1021/j100560a010
Lietti L., Nova I., Ramis G. et al. // J. Catal. 1999. V. 187. № 2. P. 419. https://doi.org/10.1006/jcat.1999.2603
Marciel A., Graça M., Bastos A. et al. // Mater. 2021. V. 14. № 4. P. 821. https://doi.org/10.3390/ma14040821
Sen U.K., Mitra S. // RSC Adv. 2012. V. 2. P. 11123. https://doi.org/10.1039/C2RA21373G
Guidi V., Cardinali G., Dori L. et al. // Sens. Actuators B Chem. 1998. V. 49. P. 88. https://doi.org/10.1016/S0925-4005(98)00039-2
Shahram Sh., Daniel V.O., Fey T. et al. // Mater. Sci. Eng. C. 2016. V. 58. P. 1064. https://doi.org/10.1016/j.msec.2015.09.069
Pershina V., Fricke B. // Russ. J. Phys. Chem. 1995. V. 99. № 1. P. 144.
Pershina V., Fricke B. // Ibid. 1996. V. 100. № 21. P. 8748.
CRC Handbook of Chemistry and Physics. 102 ed. 2021-2022: CRC Press, Taylor & Francis Group.
Elam J.W., Groner M.D., George S.M. // Rev. Sci. Instrum. 2002. V. 73. № 8. P. 2981–2987. https://doi.org/10.1063/1.1490410
Кутчиев А.И. Синтез и квантово-химическое исследование ванадийоксидных структур на поверхности кремнезема и их взаимодействия с парами VOCl3 и H2O: Дис. … канд. хим. наук. Санкт-Петербург, 2006. 171 с.
Wind R.A., George S.M. // J. Phys. Chem. A. 2010. V. 114. № 3. P. 1281. https://doi.org/10.1021/jp9049268
Wind R.W., Fabreguette F.H., Sechrist Z.A. et al. // J. Appl. Phys. 2009. V. 105. № 7. P. 074309. https://doi.org/10.1063/1.3103254
Malygin A.A., Volkova A.N., Kol’tsov S.I., Alekskovskii V.B. // Russ. J. Gen. Chem. 1972. V. 42. № 11. P. 2373.
Malygin A.A., Volkova A.N., Kol’tsov S.I., Alekskovskii V.B. // Ibid. 1973. V. 43. № 7. P. 1436.
Malygin A.A. // Ibid. 2002. V. 72. № 4. P. 575.
Ефимов А.И., Белокурова Л.П., Василькова И.В., Чечев В.П. Свойства неорганических соединений. Справочник. Л.: Химия, 1983. С. 392.
Mackus A.J.M., Schneider J.R., MacIsaac C. et al. // Chem. Mater. 2019. V. 31. № 4. P. 1142. https://doi.org/10.1021/acs.chemmater.8b02878
George S.M. // Acc. Chem. Res. 2020. V. 53. № 6. P. 1151. https://doi.org/10.1021/cr900056b
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии