

Журнал физической химии, 2022, T. 96, № 4, стр. 502-509
Спектры фотоиндуцированного поглощения донорно-акцепторных систем на основе фталоцианинов кобальта(II) и марганца(III) с фемтосекундным временным разрешением
Е. Н. Овченкова a, Н. Г. Бичан a, *, Т. Н. Ломова a
a Институт химии растворов им. Г.А. Крестова РАН
Иваново, Россия
* E-mail: bng@isc-ras.ru
Поступила в редакцию 13.10.2021
После доработки 13.10.2021
Принята к публикации 14.10.2021
- EDN: JJGWGA
- DOI: 10.31857/S0044453722040252
Аннотация
Представлены результаты исследования сверхбыстрой динамики возбужденных состояний (октакис-3,5-ди-трет-бутилфенокси)фталоцианинато)кобальта(II) и марганца(III), а также донорно-акцепторных систем на их основе с фуллеро[60]- и фуллеро[70]пирролидинами в качестве акцепторов. Возбужденные состояния достигались при воздействии на исследуемые поглощающие вещества лазерного импульса в методе фемтосекундной абсорбционной спектроскопии. Описана кинетика и виды спектров фотоиндуцированного поглощения металлофталоцианинов и их диад с замещенными пирролидинами, определены времена жизни состояний с разделенными зарядами и определены константы разделения и рекомбинации зарядов.
Донорно-акцепторные (D–A) комплексы, фрагменты которых подбираются таким образом, чтобы сформировать донорно-акцепторную пару, являются перспективными в практическом отношении фотоактивными системами. В качестве распространенных D–A-пар изучаются порфирин-фуллерены и фталоцианин-фуллерены, в которых порфирины и фталоцианины выступают в роли электронодоноров [1–5], а фуллерены – в роли акцепторов [6]. Эта роль связана с наличием системы сопряженных связей, низким потенциалом окисления и значительным поглощением в видимой области спектра у первых и уникальной трехмерной структуры с низким потенциалом восстановления и небольшой энергией реорганизации – у вторых. Применение таких D–A-систем в области создания элементов солнечной энергетики требует наличия в них эффективного фотоиндуцированного переноса электронов и долгоживущего состояния с разделенными зарядами, которое достигается за счет быстрого разделения (CS) и медленной рекомбинации зарядов (CR). Лучшим методом изучения динамики процессов и определения времен жизни состояния с разделенными зарядами являются методы исследования с временным разрешением, в частности наносекундная и фемтосекундная импульсная лазерная спектроскопия. Воздействие нано- или фемтосекундных лазерных импульсов на D–A-системы позволяет создать высокую концентрацию молекул в возбужденном состоянии с изменением их донорно-акцепторных свойств.
Быстрые процессы в порфириновых молекулах всегда интересовали исследователей и первые работы по изучению этих процессов были выполнены еще до изобретения метода фемтосекундной спектроскопии [7]. С появлением метода порфирины и фталоцианины стали объектами исследования, активно изучающимися по настоящее время [8, 9]. Большое внимание уделяется изучению фемтосекундной спектроскопии поглощения D–A-систем на их основе [10, 11]. Установлено, что эффективность фотоиндуцированного переноса электрона в порфирин- и фталоцианин-фуллереновых системах зависит от различных факторов, а именно от природы замещения в макроциклическом фрагменте [12, 13], геометрии [14] и длины [15] сопряженного мостика между D и A, структуры модифицированного фуллерена [16] и способа его связывания с донором [17]. Кроме того, фотоиндуцированный перенос электронов может быть настроен с помощью координации макрогетероциклического соединения с ионом металла. Анализ литературы показал, что большинство работ посвящено синтезу D–A-систем на основе порфиринов и фталоцианинов цинка [3, 10, 12]. Однако для расширения возможностей дизайна фотоактивных систем необходимо уделить внимание порфириновым и фталоцианиновым комплексам других d-металлов, а также s- и p-металлов. Так, например, комплексы кобальта, молибдена, рутения, обладая высокой координационной емкостью, образуют D–A-системы различного состава [18–22]. Наличие гидроксидных лигандов в порфиринах и фталоцианинах алюминия и олова открывает возможности получения ковалентных фуллерен-содержащих аксиальных комплексов [23, 24]. Координация марганца с макроциклическим лигандом также позволяет получать D–A-системы с потенциалом в качестве средств для сбора света во всем видимом и ближнем ИК-диапазоне для фотоэлектрических приложений [25–27].
Для разработки подходов в синтезе перспективных фотоактивных компонентов на основе порфирин- и фталоцианин-фуллереновых комплексов необходимо выявление взаимосвязи между химической структурой и параметрами, влияющими на эффективность процесса фотоиндуцированного переноса электрона. В настоящей работе в качестве объектов исследования выбраны D–A-системы на основе (октакис-3,5-ди-трет-бутилфенокси)фталоцианинато)кобальта(II) и марганца(III) (CoPc и (AcO)MnPc) и 1-N-метил-2-(пиридин-4-ил)-3,4-фуллеро[60 ] пирролидина (PyC60), 1-N-метил-4-(1H-имидазол-1'-ил)-3,4-фуллеро[60 ] пирролидина (ImC60) и 2,5-ди-(пиридин-2-ил)-3,4-фуллеро[70 ] пирролидина (Py2C70) (рис. 1). Для определения времен жизни состояний с разделенными зарядами использован метод фемтосекундной абсорбционной спектроскопии как актуальный при исследовании динамики процессов, время протекания которых менее 1 нс.

ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Метод получения координационных донорно-акцепторных диад
Синтез D–A-систем (координационных диад) проведен путем самосборки в системах (октакис-3,5-ди-трет-бутилфенокси)фталоцианин кобальта(II) или марганца(III) – пиридил- или имидазолил-замещенный фуллеро[60 ] - или фуллеро[70 ] пирролидин в толуоле. Подробное описание механизмов их образования, кинетических и термодинамических параметров и спектральных методов определения структуры представлено в работах [10, 28, 29]. Максимумы Q-полосы в электронных спектрах поглощения (ЭСП) и константы устойчивости как ключевые параметры диад приведены в табл. 1.
Дифференциальные спектры поглощения тетрапиррольных комплексов и донорно-акцепторных диад на их основе
Использована установка для регистрации спектров фотоиндуцированного поглощения в фемтосекундном диапазоне, созданная в Федеральном исследовательском центре химической физики им. Н.Н. Семенова РАН (Москва). Разрешенные во времени дифференциальные спектры наведенного поглощения ΔA(λ, t) измерялись методом ”возбуждение–зондирование“ с помощью фемтосекундной абсорбционной установки [10, 29] с использованием фемтосекундного титан-сапфирового лазера (800 нм, 80 МГц, 30 фс, “Цунами”, Spectra-Physics, США). Для возбуждения объектов исследования использовались усиленные импульсы (800 нм, 100 Гц, 1.5 мДж, 40 фс), которые разделялись на два пучка. Один из пучков ослабляли до энергии 0.8 мДж и направляли на неколлинеарный оптический параметрический усилитель № 1 (“NOPA”, Clark-MXR, США), излучение которого использовалось в качестве импульса накачки. Импульс накачки имел гауссову форму с максимумом излучения 690 (для CoPc и диад на его основе) и 735 нм (для (AcO)MnPc и диад на его основе), длительностью 25 фс, с энергией 150 мДж. Эта энергия является оптимальной для получения сигнала достаточной амплитуды. Второй пучок ослабляли до 0.4 мДж и направляли на неколлинеарный оптический параметрический усилитель № 2 (“Topas white”, Light Conversion, Литва). Для создания зондирующего импульса суперконтинуума излучение пучка (890 нм, 35 фс, 10 мкДж) ослаблялось до 1 мкДж и фокусировалось на тонкой кварцевой ячейке с чистой H2O. Импульсы накачки и белого континуума фокусировались в пятно диаметром 300 и 120 мкм соответственно. Поляризации импульсов накачки и зондирования были ориентированы под магическим углом 54.7° друг к другу. Суперконтинуум диспергировался с помощью полихроматора (Action SP300, Roper Scientific, США) и регистрировался с помощью CCD-камеры (“Newton”, Andor, США).
Дифференциальные спектры поглощения ΔA(λ, t) = A(λ, t) – A0(λ), возникающие при импульсном фотовозбуждении, являются разностью двух спектров исследуемых образцов: A(λ, t) при времени задержки t и исходного спектра поглощения раствора образца без возбуждения A0(λ). Дифференциальные спектры ΔA(λ, t) были записаны в спектральном диапазоне 380–850 нм. Измеренные спектры подвергались коррекции, учитывающей дисперсию групповой скорости континуума, по процедуре, описанной в работах [30, 31]. Эксперименты выполнялись при температуре 298 K в 0.5 мм оптической кювете в атмосфере азота, чтобы избежать контакта раствора пробы с воздухом.
Анализ временных характеристик дифференциальных спектров осуществлялся с помощью кинетического моделирования на основе сингулярного разложения (SVD) матрицы полученных данных. Используемый подход предполагает рассмотрение всего массива экспериментальных значений ΔA(λ, t), где ΔA отображает фотоиндуцированные изменения оптической плотности исследуемого образца (просветление и наведенное поглощение). Экспоненциальная обработка полученных данных на длине волны возбужденного состояния использована для определения времени жизни возбужденного состояния и констант скорости процессов переноса электрона.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
ЭСП CoPc и (AcО)MnPc имеют интенсивные Q-полосы при 673 и 727 нм, соответственно, обусловленные переходами ВЗМО–НВМО (a1u → → eg). Высокая интенсивность Q-полос фталоцианиновых комплексов относительно макрогетероциклических аналогов, например металлопорфиринов, связана с расширением цикла и заменой четырех Cмезо- на N-атомы, что нарушает энергетический баланс между занятыми граничными орбиталями (a1u и a2u) [32]. При образовании донорно-акцепторных комплексов исследуемых фталоцианинов c фуллеро[60 ] - и фуллеро[70 ] -пирролидинами Q-полосы в их ЭСП батохромно смещаются на 3–5 нм (табл. 1). Наблюдается уменьшение интенсивности Q-полос для всех случаев в 1.2–2 раза, что связано с появлением дополнительной донорно-акцепторной связи M–Nфуллерен, которая вносит вклад в баланс энергетических уровней.
Дифференциальные спектры поглощения CoPc и (AcO)MnPc, измеренные в спектральном диапазоне от 380 до 850 нм при определенной временной задержке, представлены на рис. 2.
Рис. 2.
Дифференциальные спектры поглощения CoPc (а) и (AcO)MnPc (б) при возбуждении импульсом 690 нм (а) и 735 нм (б) в толуоле, насыщенном азотом. На вставках показаны соответствующие кинетические кривые.
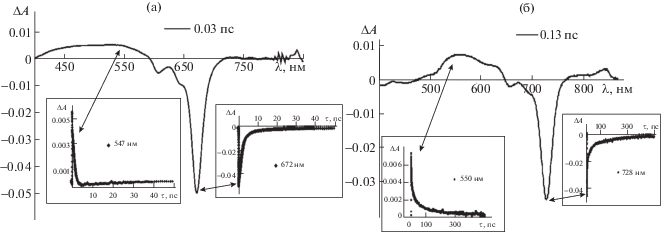
В дифференциальных спектрах можно выделить полосы поглощения ∆А(λ, t) > 0 из электронно-возбужденных состояний (excited state absorption – ESA-полосы) и полосы выцветания ∆А(λ, t) < < 0 (bleaching – BL-полосы) [33]. BL-полосы в дифференциальных спектрах обусловлены обеднением основного состояния комплексов за счет возбуждения. В дифференциальном спектре поглощения CoPc (рис. 2а) BL полосы проявляются при 608, 643 и 672 нм в области Q-полосы, а ESA-полоса простирается от 478 до 550 нм. Вставка на рис. 2а показывает, что относительная интенсивность пика выцветания при 672 нм существенно изменяется во времени, тогда как изменение интенсивности пика возбужденного состояния при 547 нм менее значительно. Временной профиль для полос при 672 и 547 нм показывает очень быстрое восстановление этих сигналов. Постоянная времени для восстановления сигнала при 672 нм составляла 2.14 пс, в то время как экспоненциальное затухание пика при 547 нм показало постоянную времени 1.05 пс (рис. 2а, вставка).
В случае (AcO)MnPc (рис. 2б) полосы выцветания проявляются при 657 и 728 нм, тогда как полосы возбужденного состояния проявляются в области 520–630 нм и при 845 нм. Временной профиль поглощения (AcO)MnPc при 550 нм (вставка на рис. 2б) хорошо описывается триэкспоненциальной функцией, показывая два коротких времени жизни 0.68 и 15.89 пс и заметную составляющую длительного времени жизни 111.24 пс. Таким образом, замена атома металла во фталоцианиновом комплексе способствует изменению времени жизни возбужденного состояния.
Фотоиндуцированный перенос электрона от молекул CoPc и (AcO)MnPc к фуллерен-содержащему аксиальному лиганду наблюдается при возбуждении диад, растворенных в деаэрированном толуоле, импульсным лазерным излучением с длиной волны 690 и 735 нм соответственно.
Дифференциальные спектры поглощения на примере (PyC60)CoPc и (AcO)(PyC60)MnPc, измеренные в спектральном диапазоне от 380 до 850 нм при различных временах задержки, представлены на рис. 3 и 4. В дифференциальных спектрах поглощения диад наблюдаются небольшие сдвиги полос, подтверждающие образование состояния с разделенными зарядами – ${\text{CoP}}{{{\text{c}}}^{{ \bullet + }}}$ : ${\text{PyC}}_{{60}}^{{ \bullet - }}$ и (AcO)${\text{MnP}}{{{\text{c}}}^{{ \bullet + }}}$ : ${\text{PyC}}_{{60}}^{{ \bullet - }}$. Так, в случае (PyC60)CoPc BL полосы проявляются при 610, 644 и 677 нм, а ESA полосы (полосы ${\text{CoP}}{{{\text{c}}}^{{ \bullet + }}}$) – при 548 и 729 нм (табл. 2). В случае (AcO)(PyC60)MnPc полосы выцветания находятся при 656 и 727 нм, а полосы возбужденного состояния (полосы (AcO)${\text{MnP}}{{{\text{c}}}^{{ \bullet + }}}$) – при 561 и 835 нм (табл. 2). К сожалению, условия эксперимента (спектральный диапазон приборной установки от 400 до 900 нм) не позволяют обнаружить переходную полосу π-анион радикальной формы ${\text{PyC}}_{{60}}^{{ \bullet - }}$ в диадах, так как она проявляется в ближней ИК-области от 950 до 1020 нм [16, 34].
Рис. 3.
Дифференциальные спектры поглощения (PyC60)CoPc при возбуждении импульсом 690 нм в деаэрированном толуоле. На вставке показана соответствующая кинетическая кривая.

Рис. 4.
Дифференциальные спектры поглощения (AcO)(PyC60)MnPc при возбуждении импульсом 735 нм в деаэрированном толуоле. На вставке показана соответствующая кинетическая кривая.

Таблица 2.
Максимумы BL- и ESA-полос в дифференциальных спектрах поглощения диад, длина волны поглощения с временным профилем (λ, нм), времена образования и жизни возбужденного состояния (τCS, τCR) и константы скорости разделения (kCS) и рекомбинации (kCR) зарядов в диадах в толуоле
| Комплекс | BL-полосы, нм | ESA-полосы, нм | λ, нм | τCS, пс | kCS, с–1 | τCR, пс | kCR, с–1 |
|---|---|---|---|---|---|---|---|
| CoPc | 608, 643, 672 | в области 478–550 | 547 | 0.003 | 1.05 | ||
| (PyC60)CoPc | 610, 643, 677 | 548, 729 | 548 | 0.005 | 2.0 × 1014 | 1.05 9.92 |
9.52 × 1011 1.01 × 1011 |
| (ImC60)CoPc | 611, 643, 677 | 548, 840 | 548 | 0.007 | 1.43 × 1014 | 1.55 20.89 |
6.45 × 1011 4.78 × 1010 |
| (AcO)MnPc | 657, 728 | в области 520−630, 845 | 550 | 0.03 | 0.68 15.89 111.24 |
||
| (AcO)(PyC60)MnPc | 656, 727 | 561, 835 | 561 | 0.06 | 1.67 × 1013 | 0.30 16.84 144.41 |
3.33 × 1012 5.94 × 1010 6.92 × 109 |
| (AcO)(ImC60)MnPc | 655, 728 | 557, 838 | 557 | 0.074 | 1.35 × 1013 | 0.38 16.74 128.71 |
2.63 × 1012 5.97 × 1010 7.77 × 109 |
| (AcO)(Py2C70)MnPc | 658, 734 | 553, 837 | 553 | 0.085 | 1.18 × 1013 | 0.39 8.63 82.63 |
2.56 × 1012 1.16 × 1011 1.24 × 1010 |
Время жизни состояния с разделенными зарядами в D–A-диадах определялось путем экспоненциальной аппроксимации экспериментальных кривых дифференциального поглощения (рис. 3, 4, вставки). Первый участок кинетической кривой описывает рост сигнала разностного поглощения и позволяет определить время образования возбужденного состояния (τCS), а второй участок, характеризующийся падением разностного поглощения, позволяет определить его время жизни (τCR). Обнаружено, что возбужденное состояние ${\text{CoP}}{{{\text{c}}}^{{ \bullet + }}}$ в диадах (PyC60)CoPc и (ImC60)CoPc образуется за 5 и 7 фс и его время жизни, определенное по двухэкспоненциальной зависимости, составляет 9.92 и 20.89 пс соответственно. В случае диад на основе марганец(III) фталоцианина время образования возбужденного состояния (AcO)${\text{MnP}}{{{\text{c}}}^{{ \bullet + }}}$ около 0.06–0.08 пс, а время его жизни описывается трехэкспоненциальной зависимостью и самая длительная составляющая τCR равна 144.41, 128.71 и 82.63 пс для (AcO)(PyC60)MnPc, (AcO)(ImC60)MnPc и (AcO)(Py2C70)MnPc соответственно (табл. 2). Константы скорости разделения (kCS) и рекомбинации (kCR) зарядов представлены в табл. 2.
Анализ данных табл. 2 показывает, что величина kCR для D–A-диад на 1–3 порядка меньше, чем значения kCS. Такая тенденция обычно наблюдается для порфирин- и фталоцианин-фуллеренновых диад из-за низкой энергии реорганизации фуллерена [12, 16]. Из табл. 2 также видно, что вариация фуллеропирролидинов в составе D–A-диад влияет на значения kCR. По результатам определения времен жизни состояний с разделенными зарядами, τCR, системы на основе (AcO)MnPc более перспективны для оптоэлектроники по сравнению с таковыми на основе CoPc.
Несмотря на то, что результаты фемтосекундных исследований показывают малое время жизни возбужденных состояний порфиринов и фталоцианинов кобальта(II) [29, 35, 36], они могут использоваться в качестве донорных строительных блоков для систем с переносом заряда, построенных по другим стратегиям. В работах [37, 38] методом самосборки в растворе были получены сокристаллы с переносом заряда на основе порфирина кобальта и фуллеренов (C70, C60), которые демонстрируют превосходную электропроводность, высокую “подвижность” дырок, чрезвычайно высокую термическую стабильность и сильные межмолекулярные взаимодействия между фуллереном и порфирином кобальта. В работах [37, 38] также было показано, что сокристаллизация с переносом заряда является мощной стратегией для рационального проектирования и создания широкого класса проводящих элементов для электроники нового поколения. Авторы работ [39, 40] для выяснения причин повышенной электрокаталитической активности систем на основе оксида графена и порфирина кобальта исследовали их, используя спектроскопические измерения с временным разрешением и теоретические расчеты. Фотовозбуждение этих наногибридных систем видимым светом приводит к быстрому разделению зарядов, вызывая восстановление электроноакцепторных одностенных углеродных нанотрубок и окисление электронодонорных порфиринов. Измерения переходного поглощения подтверждают, что ион-радикальные пары являются долгоживущими, со временем жизни в микросекундном диапазоне. В работе [36] показано, что время жизни возбужденного состояния порфирина кобальта(II) может быть увеличено изменением природы растворителя. Мы провели исследование фемтосекундных спектров CoPc в деаэрированном 1,2-дихлорбензоле. CoPc демонстрирует хорошую растворимость и стабильность при сильном лазерном облучении в этой среде, как и в толуоле. В дифференциальном спектре CoPc в 1,2-дихлорбензоле полосы выцветания проявляются при 612, 649 и 678 нм, а полосы возбужденного состояния при 471 и 713 нм (рис. 5). Время образования возбужденного состояния, определенное из временного профиля поглощения при 713 нм, составляет 1.52 пс, а время его жизни 26.86 пс. Таким образом, замена толуола на 1,2-дихлорбензол в качестве растворителя увеличило τCR в 26 раз.
Рис. 5.
Дифференциальные спектры поглощения CoPc при возбуждении импульсом 685 нм в деаэрированном 1,2-дихлорбензоле. На вставках показаны соответствующие кинетические кривые.

Таким образом, в данной работе методом фемтосекундной абсорбционной спектроскопии показано, что октакис-3,5-ди-трет-бутилфенокси)фталоцианины кобальта(II) и марганца(III) образуют фотоиндуцированные состояния с разделенными зарядами – радикальные соли в составе донорно-акцепторных координационных диад с N-гетероциклическими производными фуллеро[60 ] - и фуллеро[70 ] пирролидинами. Анализом дифференциальных спектров и временных профилей поглощения D–A-диад получены ключевые константы для их возбужденных состояний в зависимости от природы иона металла во фталоцианиновом доноре электрона, строения фуллерен-содержащего акцептора и растворителя. Так, на количественном уровне показано, что фталоцианины кобальта(II) и марганца(III) пригодны для получения D–A-систем с акцепторами фуллереновой природы и что их возбужденными состояниями в виде радикальных солей можно управлять.
Работа выполнена при поддержке Российского научного фонда, грант РНФ № 21-73-20090. Авторы выражают благодарность за проведение исследований дифференциальных спектров поглощения донорно-акцепторных систем проф., д.х.н. В.А. Надточенко, к.х.н. И.В. Шелаеву, Ф.Е. Гостеву. Работа выполнена на оборудовании Федерального исследовательского центра химической физики им. Н.Н. Семенова Российской академии наук (Москва) и Центра коллективного пользования научным оборудованием “Верхневолжский региональный центр физико-химических исследований”.
Список литературы
Zhu P.H., Song F.F., Ma P. et al. // Dyes Pigments. 2018. V. 151. P. 385. https://doi.org/10.1016/j.dyepig.2018.01.012
Бичан Н.Г., Овченкова Е.Н. // Изв. АН, Сер. хим. 2021. Т. 70. № 2. С. 239. https://doi.org/10.1007/s11172-021-3081-y
Lu X.-J., Zhang C.-R., Shen Y.-L. et al. // J. Mol. Struct. 2018. V. 1173. P. 398. https://doi.org/10.1016/j.molstruc.2018.07.018
Ji W., Wang T.-X., Ding X. et al. // Coord. Chem. Rev. 2021. V. 439. P. 213875. https://doi.org/10.1016/j.ccr.2021.213875
Basiuk V.A., Tahuilan-Anguiano D.E. // Chem. Phys. Lett. 2019. V. 722. P. 146. https://doi.org/10.1016/j.cplett.2019.03.019
Ganesamoorthy R., Sathiyan G., Sakthivel P. // Sol. Energy Mater. Sol. Cells. 2017. V. 161. P. 102. https://doi.org/10.1016/j.solmat.2016.11.024
Schwarz F.P., Gouterman M., Muljiani Z. et al. // Department of Chemistry, Harvard University, Cambridge Bioinorg. Chem. 1972. V. 2. № 1. P. 1.
Bhattacharya S., Reddy G., Paul S. et al. // Dyes Pigments. 2021. V. 184. P. 108791. https://doi.org/10.1016/j.dyepig.2020.108791
Hong J., Fauvell T.J., Helweh W. et al. // J. Photochem. Photobiol. A: Chem. 2019. V. 372. P. 270. https://doi.org/10.1016/j.jphotochem.2018.12.026
Ovchenkova E.N., Bichan N.G., Gostev F.E. et al. // Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2021. V. 263. P. 120166. https://doi.org/10.1016/j.saa.2021.120166
Kc C.B., D’Souza F. // Coord. Chem. Rev. 2016. V. 322. P. 104. https://doi.org/10.1016/j.ccr.2016.05.012
Das S.K., Song B., Mahler A. et al. // J. Phys. Chem. C. 2014. V. 118. № 8. P. 3994. https://doi.org/10.1021/jp4118166
Follana-Berná J., Seetharaman S., Martín-Gomis L. et al. // Phys. Chem. Chem. Phys. 2018. V. 20. № 11. P. 7798. https://doi.org/10.1039/C8CP00382C
Wijesinghe C.A., El-Khouly M.E., Zandler M.E. et al. // Chem. – A Eur. J. 2013. V. 19. № 29. P. 9629. https://doi.org/10.1002/chem.201300877
Pelado B., Abou-Chahine F., Calbo J. et al. // Chem. – A Eur. J. 2015. V. 21. № 15. P. 5814. https://doi.org/10.1002/chem.201406514
Seetharaman S., Jang Y., Kc C. et al. // J. Porphyrins Phthalocyanines. 2018. V. 21. № 12. P. 1. https://doi.org/10.1142/s1088424617500924
Ince M., Hausmann A., Martinez-Diaz M.V. et al. // Chem. Commun. 2012. V. 48. № 34. P. 4058. https://doi.org/10.1039/c2cc30632h
Rodríguez-Morgade M.S., Plonska-Brzezinska M.E., Athans A.J. et al. // J. Am. Chem. Soc. 2009. V. 131. № 30. P. 10484. https://doi.org/10.1021/ja902471w
Motorina E.V., Lomova T.N., Klyuev M.V. // Mendeleev Commun. 2018. V. 28. № 4. P. 426. https://doi.org/10.1016/j.mencom.2018.07.029
Bichan N.G., Ovchenkova E.N., Mozgova V.A. et al. // Polyhedron. 2021. V. 203. P. 115223. https://doi.org/10.1016/j.poly.2021.115223
Ovchenkova E.N., Bichan N.G., Gruzdev M.S. et al. // New J. Chem. 2021. V. 45. P. 9053. https://doi.org/10.1039/D1NJ00980J
Subedi D.R., Jang Y., Ganesan A. et al. // J. Porphyrins Phthalocyanines. 2021. V. 25. № 05–06. P. 533. https://doi.org/10.1142/S1088424621500449
Zarrabi N., Poddutoori P.K. // Coord. Chem. Rev. 2021. V. 429. P. 213561. https://doi.org/10.1016/j.ccr.2020.213561
Zhao H., Zhu Y., Chen C. et al. // Polymer. 2014. V. 55. № 8. P. 1913. https://doi.org/10.1016/j.polymer.2014.02.058
El Mahdy A.M., Halim S.A., Taha H.O. // J. Mol. Struct. 2018. V. 1160. P. 415. https://doi.org/10.1016/j.molstruc.2018.02.041
Ovchenkova E.N., Bichan N.G., Kudryakova N.O. et al. // Dyes Pigments. 2018. V. 153. № P. 225. https://doi.org/10.1016/j.dyepig.2018.02.023
Овченкова Е.Н., Бичан Н.Г., Ломова Т.Н. // Журн. орган. химии. 2016. Т. 52. № 10. С. 1509. https://doi.org/10.1134/s1070428016100213
Bichan N.G., Ovchenkova E.N., Tsaturyan A.A. et al. // New J. Chem. 2020. V. 44. № 26. P. 11262. https://doi.org/10.1039/D0NJ02166K
Ovchenkova E.N., Bichan N.G., Tsaturyan A.A. et al. // J. Phys. Chem. C. 2020. V. 124. № 7. P. 4010. https://doi.org/10.1021/acs.jpcc.9b11661
Shelaev I.V., Gostev F.E., Vishnev M.I. et al. // J. Photochem. Photobiol. B: Biology. 2011. V. 104. № 1. P. 44. https://doi.org/10.1016/j.jphotobiol.2011.02.003
Kovalenko S.A., Dobryakov A.L., Ruthmann J. et al. // Phys. Rev. A. 1999. V. 59. № 3. P. 2369. https://doi.org/10.1103/PhysRevA.59.2369
Mack J., Stillman M.J., in “The Porphyrin Handbook” (Kadish K.M., Smith K.M., and Guilard R., eds.). Academic Press. Amsterdam, 2003. P. 43.
Михайлов К.М. Динамика фотовозбужденных димеров и тримеров порфирината цинка на фемто- и пикосекундной шкале времен: Дис. … канд. хим. наук. М.: Институт химической физики РАН, 2018. 97 с.
Guldi D.M., Rahman G.M.A., Jux N. et al. // J. Am. Chem. Soc. 2005. V. 127. № 27. P. 9830. https://doi.org/10.1021/ja050930o
Loppnow G.R., Melamed D., Leheny A.R. et al. // J. Phys. Chem. 1993. V. 97. № 35. P. 8969. https://doi.org/10.1021/j100137a022
Yu H.Z., Baskin J.S., Steiger B. et al. // Chem. Phys. Lett. 1998. V. 293. № 1. P. 1. https://doi.org/10.1016/S0009-2614(98)00753-2
Zheng S., Zhong J., Matsuda W. et al. // Nano Research. 2018. V. 11. № 4. P. 1917. https://doi.org/10.1007/s12274-017-1809-7
Wakahara T., D’Angelo P., Miyazawa K.I. et al. // J. Am. Chem. Soc. 2012. V. 134. № 17. P. 7204. https://doi.org/10.1021/ja211951v
Cho S., Lim J.M., You J.-M. et al. // Isr. J. Chem. 2016. V. 56. № 2–3. P. 169. https://doi.org/10.1002/ijch.201500022
You J.-M., Han H.S., Lee H.K. et al. // Int. J. Hydrogen Energy. 2014. V. 39. № 10. P. 4803. https://doi.org/10.1016/j.ijhydene.2014.01.107
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии