

Физика металлов и металловедение, 2021, T. 122, № 7, стр. 713-720
Структура и энергия симметричных границ наклона с осью 〈110〉 в Ni и энергии образования вакансий в границах зерен
М. Г. Уразалиев a, М. Е. Ступак a, В. В. Попов a, *
a Институт физики металлов им. М.Н. Михеева УрО РАН
620108 Екатеринбург, ул. С. Ковалевской, 18, Россия
* E-mail: vpopov@imp.uran.ru
Поступила в редакцию 15.12.2020
После доработки 16.02.2021
Принята к публикации 04.03.2021
Аннотация
Методами компьютерного моделирования с использованием потенциала погруженного атома исследовано четырнадцать симметричных границ наклона с осью 〈110〉 в Ni. Методом молекулярно-статического моделирования рассчитаны структуры, энергии и ширина границ зерен. Показано, что структура симметричных границ наклона с осью 〈110〉 может быть представлена ограниченным числом структурных элементов. Устойчивость структур границ при повышенной температуре исследована методом молекулярной динамики. Рассчитаны энергии образования вакансий в границах зерен.
ВВЕДЕНИЕ
Атомная структура и энергия границ зерен (ГЗ) и энергия образования в них точечных дефектов уже много лет вызывают повышенный интерес исследователей. Это обусловлено тем, что эти параметры существенно влияют на многие свойства поликристаллических материалов [1]. Прямое экспериментальное определение этих параметров связано со значительными трудностями. Поэтому широкое распространение получило атомистическое моделирование структуры и свойств ГЗ. Несмотря на то, что Ni имеет широкое промышленное применение, имеется относительно мало работ, в которых бы моделировалась структура и рассчитывалась энергия границ зерен в нем. При этом в большинстве работ расчеты выполнены только для каких-то отдельных границ, например, [2, 3], а в работе [4], в которой расчеты выполнены для широкого диапазона углов разориентировки, использовался потенциал из работы [5], который дает очень низкое значение энергии дефектов упаковки, что приводит к диссоциации границ.
Целью настоящей работы являлось моделирование структуры и оценка энергии симметричных границ наклона с осью 〈110〉 в поликристаллическом Ni, а также оценка энергии образования вакансий в таких границах. В настоящей работе ставилась задача провести систематический анализ структуры и энергии границ зерен в Ni в широком диапазоне углов разориентировки. Кроме того, ставилась задача оценить энергию образования вакансий в этих границах.
МЕТОДОЛОГИЯ АТОМИСТИЧЕСКОГО МОДЕЛИРОВАНИЯ
В настоящей работе методом компьютерного моделирования были исследованы симметричные границы наклона осью 〈110〉 в Ni, охватывающие весь диапазон углов наклона. С использованием программы Lammps [6] были выполнены расчеты для 14 большеугловых границ с осью наклона 〈110〉:
Идеальные ориентации кристаллов, соответствующие границам Σ = 1, (001), 0° и Σ = 1, (110), 180° включены в качестве конечных точек этого диапазона углов наклона.
Расчетная ячейка и граничные условия. Расчетная ячейка содержала два блока с периодическими граничными условиями Борна–Кармана в трех ортогональных направлениях. Процедура построения симметричных ГЗ была аналогична примененной в работах [7, 8]. Для создания специальной границы зерен каждый блок моделирования разворачивали один относительно другого на угол, соответствующий специальному углу согласно теории решеток совпадающих узлов.
Ячейка моделирования имеет две идентичных ГЗ: ГЗ1, повторяющаяся на верхней и нижней границах ячейки вследствие периодических граничных условий, и ГЗ2 между двумя блоками (зернами) (рис. 1).
Рис. 1.
Схема трехмерной периодической бикристаллической расчетной ячейки с обозначением ориентации кристалла. с – ось наклона, n – нормаль к плоскости границы [7].
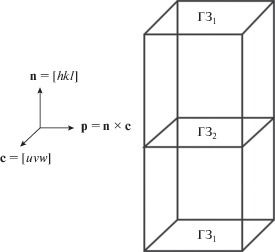
Направляющие вектора блоков были следующие: нормаль к плоскости границы зерна, n, вектор оси наклона, с, вектор p, равный векторному произведению векторов нормали к плоскости границы и оси наклона.
На этапе построения ГЗ соседние зерна могли содержать атомы, которые расположены очень близко друг к другу (ближе удвоенного атомного радиуса). В этом случае атом одного из соседних зерен удалялся. После удаления атомов выполняли редактирование геометрии блоков относительно друг друга. Тем самым задавали разные начальные конфигурации. Подобная процедура использовалась в работах [7, 8]. Размер блока моделирования находился в диапазоне от 2000 до 3000 атомов.
Потенциалы межатомного взаимодействия. При построении потенциалов методом погруженного атома предполагали, что функции электронной плотности радиально симметричны. Поэтому эти потенциалы лучше всего подходят для моделирования элементов, имеющих ненаправленные связи, в частности для Ni, имеющего ГЦК-кристаллическую решетку. К настоящему времени методом погруженного атома построено несколько потенциалов межатомного взаимодействия для Ni [5, 9–12]. Чтобы выбрать среди них потенциал, наилучшим образом описывающий межатомное взаимодействие, с помощью программы Lammps, используя различные потенциалы, были рассчитаны период решетки, упругие модули, энергия когезии, энергия образования вакансий и энергия дефектов упаковки в Ni и проведено сравнение полученных значений с экспериментальными (табл. 1).
Таблица 1.
Сравнение значений периода кристаллической решетки, упругих модулей, энергии когезии, энергии образования вакансий и энергии дефектов упаковки в Ni, рассчитанных с использованием различных потенциалов межатомного взаимодействия, и полученных в эксперименте
| Свойства | Расчет с использованием различных потенциалов межатомного взаимодействия | Эксперимент | ||||
|---|---|---|---|---|---|---|
| [5] | [9] | [10] | [11] | [12] | ||
| a, нм | 0.3520 | 0.3520 | 0.3520 | 0.3520 | 0.3518 | 0.3524 [13] |
| C11, ГПа | 233 | 248 | 247 | 241 | 247 | 247 [14] |
| C12, ГПа | 154 | 148 | 147 | 150 | 147 | 147 [14] |
| C44, ГПа | 128 | 125 | 125 | 127 | 123 | 125 [14] |
| Энергия когезии, эВ/атом | –4.45 | –4.45 | –4.45 | –4.45 | –4.39 | –4.45 [15] |
| Энергия образования вакансии, эВ | 1.63 | 1.60 | 1.71 | 1.57 | 1.76 | 1.60 [16] |
| Энергия дефекта упаковки, мДж/м2 | 12 | 112 | 97 | 134 | 181 | 125 [17] |
Видно, что значения периода решетки, упругих модулей, энергии когезии и энергии образования вакансий, рассчитанные с использованием всех рассматриваемых потенциалов, достаточно близки к экспериментальным значениям. Значительно хуже соответствуют экспериментальным данным расчетные значения энергии дефектов упаковки. Наиболее близкое к экспериментальному значение энергии дефектов упаковки дает расчет с использованием потенциала, полученного в работе [11]. Поэтому именно этот потенциал мы использовали в наших расчетах.
Моделирование методом молекулярной статики. Релаксированные при 0 K структуры ГЗ были найдены с помощью молекулярной статики. Молекулярная статика – это метод минимизации энергии, который находит ближайший локальный минимум энергии для данной начальной конфигурации.
Энергию ГЗ, EGB, рассчитывали с помощью выражения
где Ep – полная потенциальная энергия релаксированного бикристалла, N – число атомов в системе, Ecoh – энергия когезии (энергия связи, приходящаяся на один атом решетки идеального кристалла), S – площадь ГЗ. Наличие двух границ учитывает удвоенная площадь S в знаменателе.Структуру и энергию ГЗ для каждой разориентировки получали для различных начальных конфигураций в результате поиска локального минимума энергии, методом сопряженных градиентов. В дальнейшем анализировали структуру, соответствующую глобальному минимуму энергии.
В работе также была оценена ширина ГЗ. В качестве средства наблюдения за моделью ГЗ использовали программу OVITO [18], с помощью которой было измерено максимальное расстояние между выбранными атомами по обе стороны границы. Для измерения были выбраны атомы, для которых межатомное расстояние по крайней мере в одном направлении существенно (более, чем на 0.01 нм) отличалось от межатомного расстояния в регулярной решетке. Это расстояние принимали за ширину ГЗ.
Моделирование отжига методом молекулярной динамики. Моделирование решетки методом молекулярной статики генерирует структуру границ, которая соответствуют минимуму энергии при 0 К. Тепловые флуктуации, которые присутствуют при повышенных температурах, могут быть достаточными, чтобы позволить ГЗ релаксировать к структуре с более низкой энергией. Если это так, то это означает, что структура, построенная в результате молекулярно-статического моделирования, не имеет физического смысла, поскольку она будет нестабильной при повышенных температурах. Подобный критерий стабильности структуры ГЗ использовался в работе [4].
Чтобы проверить устойчивость структуры ГЗ, построенных методом молекулярной статики для 0 К, при конечных температурах их отжигали методом молекулярно-динамического моделирования. Стабильная структура определяется как структура, в которой положения атомов не меняются после отжига. Отжиг проводился следующим образом. Исходным состоянием была структура, смоделированная методом молекулярной статики. Далее выполнялся нагрев от 0 до 600 К в течение 100 пикосекунд. Температура отжига для выбранного потенциала соответствует приблизительно 0.35 температуры плавления. Контроль изменения температуры производили с помощью термостата Нозе–Гувера. После завершения нагревания система выдерживается в течении 100 пикосекунд при температуре 600 K, после чего запускается процедура минимизации энергии ГЗ при 0 К.
Энергия образования вакансий. Были проведены расчеты энергии образования вакансий в позициях, расположенных на разном расстоянии от плоскости границы. Энергию образования вакансий рассчитывали по формуле
(2)
${{E}_{{\text{v}}}} = {{E}_{{\text{f}}}}--({{N}_{0}}--1)({{{{E}_{i}}} \mathord{\left/ {\vphantom {{{{E}_{i}}} {{{N}_{0}}}}} \right. \kern-0em} {{{N}_{0}}}}),$Энергия образования вакансии имеет физический смысл, эквивалентный тому, как если бы это была энергия, необходимая для удаления атома из позиции в ГЗ и создания атома в идеальной решетке бесконечно далеко от ГЗ.
Энергия образования вакансии зависит от позиции вакансии в окрестности ГЗ, и поэтому может быть представлена как функция расстояния между вакансией и зернограничной плоскостью.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
На рис. 2 показаны релаксированные структуры ГЗ, исследованных в настоящей работе. Структуры границ показаны перпендикулярно оси наклона.
Рис. 2.
Релаксированные структуры рассчитанных границ зерен наклона в Ni. а – Σ = 1, (001), 0°; б – $\Sigma 33,(118)\left[ {1\bar {1}0} \right]$ θ = 20.1°; в – $\Sigma 19,(116)\left[ {1\bar {1}0} \right],$ θ = 26.5°; г – $\Sigma 9,(114)\left[ {1\bar {1}0} \right],$ θ = 38.9°; д – $\Sigma 11,(113)\left[ {1\bar {1}0} \right],$ θ = 50.5°; е – $\Sigma 3,(112)\left[ {1\bar {1}0} \right],$ θ = 70.5°; ж – $\Sigma 43,(335)\left[ {1\bar {1}0} \right],$ θ = 80.6°; з – $\Sigma 3,(111)\left[ {1\bar {1}0} \right],$ θ = 109.5°; и – $\Sigma 9,(221)\left[ {1\bar {1}0} \right],$ θ = 141.1°; к – $\Sigma 27,(552)\left[ {1\bar {1}0} \right],$ θ = 148.4°. Атомы в двух последовательных плоскостях, перпендикулярных оси наклона, показаны черным или белым цветом.

Черные и светлые кружки обозначают позиции атомов в чередующихся плоскостях перпендикулярно осям наклона. Пунктирные линии охватывают структурные элементы.
Большинство границ зерен содержат структурные элементы A, B, C и D, но встречаются и границы, содержащие другие, более сложные элементы. Это границы $\Sigma 17,(223)\left[ {1\bar {1}0} \right],$ θ = 86.6° и $\Sigma 17,(334)\left[ {1\bar {1}0} \right],$ θ = 93.4°.
Границы $\Sigma 11,(113)\left[ {1\bar {1}0} \right],$ θ = 50.5°; Σ3, (112) $\left[ {1\bar {1}0} \right],$ θ = 70.5°; $\Sigma 43(335)\left[ {1\bar {1}0} \right],$ θ = 80.6°, Σ17, (223) $\left[ {1\bar {1}0} \right],$ θ = 86.6; $\Sigma 3,(111)\left[ {1\bar {1}0} \right],$ θ = 109.5°; $\Sigma 11,(332)\left[ {1\bar {1}0} \right],$ θ = = 129.5°; $\Sigma 9,(221)\left[ {1\bar {1}0} \right],$ θ = 141.1° и $\Sigma 27,(552)\left[ {1\bar {1}0} \right],$ θ = 148.4° образованы одним типом структурных элементов. Это так называемые “предпочтительные” границы. Границы $\Sigma 11,(113)\left[ {1\bar {1}0} \right],$ θ = = 50.5° и $\Sigma 3,(111)\left[ {1\bar {1}0} \right],$ θ = 109.5° – это когерентные двойниковые границы.
Атомы в двух последовательных плоскостях, перпендикулярных оси наклона, показаны черным или серым цветом.
Представляет интерес сравнить рассчитанные структуры ГЗ для Ni и других ГЦК-металлов. Такое сравнение показывает, что структуры ГЗ для Ni практически не отличаются от структур ГЗ для Al, рассчитанных в работе [4]. Однако сравнение рассчитанных в настоящей работе структур ГЗ для Ni и структур ГЗ для Cu, рассчитанных в работе [19], показывает, что между ними имеются заметные различия. Очевидно, что такие различия обусловлены различиями в потенциалах межатомного взаимодействия.
Для всех рассмотренных ГЗ был выполнен отжиг методом молекулярной динамики, который показал, что все рассчитанные методом молекулярной статики структуры ГЗ устойчивы к тепловым флуктуациям.
Нами были рассчитаны энергия ГЗ, EGB, структурная ширина ГЗ, δ, и минимальная энергия образования вакансий в ГЗ, Ev. Эти результаты приведены в табл. 2.
Таблица 2.
Параметры структуры ГЗ, энергия, ширина ГЗ и минимальная энергия образования вакансий в ГЗ
| Σ | Плоскость границы | Ось наклона | θ, град | EGB, Дж/м2 | δ, нм | Ev, эВ |
|---|---|---|---|---|---|---|
| 33 | (118) | $[1\bar {1}0]$ | 20.1 | 1.10 | 0.55 | 0.99 |
| 19 | (116) | $[1\bar {1}0]$ | 26.5 | 1.23 | 0.54 | 0.72 |
| 9 | (114) | $[1\bar {1}0]$ | 38.9 | 1.05 | 0.50 | 1.10 |
| 11 | (113) | $[1\bar {1}0]$ | 50.5 | 0.47 | 0.24 | 1.31 |
| 3 | (112) | $[1\bar {1}0]$ | 70.5 | 1.00 | 0.48 | 1.06 |
| 43 | (335) | $[1\bar {1}0]$ | 80.6 | 1.15 | 0.51 | 1.19 |
| 17 | (223) | $[1\bar {1}0]$ | 86.6 | 1.11 | 0.52 | 1.18 |
| 17 | (334) | $[1\bar {1}0]$ | 93.4 | 1.04 | 0.53 | 0.99 |
| 3 | (111) | $[1\bar {1}0]$ | 109.5 | 0.07 | 0.40 | 1.57 |
| 11 | (332) | $[1\bar {1}0]$ | 129.5 | 1.27 | 0.52 | 0.80 |
| 9 | (221) | $[1\bar {1}0]$ | 141.1 | 1.32 | 0.58 | 0.96 |
| 27 | (552) | $[1\bar {1}0]$ | 148.4 | 1.39 | 0.66 | 0.69 |
На рис. 3 показана рассчитанная зависимость энергии границ наклона с осью 〈110〉 от угла разориентировки. Границы, отвечающие углам 0° и 180°, соответствуют Σ1, и для них энергии ГЗ равны нулю. На зависимости энергии границы зерна от угла разориентировки имеются два глубоких минимума при θ = 50.5° и θ = 109.5°, соответствующих двойниковым границам $\Sigma 11,(113)\left[ {1\bar {1}0} \right]$ и $\Sigma 3,(111)\left[ {1\bar {1}0} \right],$ соответственно.
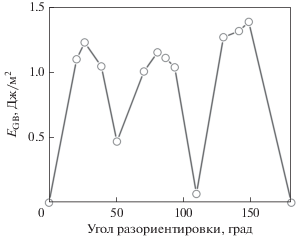
Данная зависимость близка к зависимости, полученной в работе [4]. При этом нужно отметить, что в работе [4] при расчетах использовался потенциал из работы [5], который давал очень низкую энергию дефектов упаковки. Следствием этого была диссоциация границ зерен, что приводило к изменению их структуры и некоторому уменьшению зернограничной энергии.
Нами было проведено сравнение полученных значений зернограничной энергии с результатами первопринципных расчетов, выполненных в работе [20]. В этой работе были рассчитаны энергии границ $\Sigma 11,(113)\left[ {1\bar {1}0} \right]$ и $\Sigma 3,(111)\left[ {1\bar {1}0} \right]$ и получены значения 0.47 и 0.09 Дж/м2, соответственно, которые практически совпадают со значениями, полученными в настоящей работе.
Также было проведено сравнение энергий границ зерен, рассчитанных в настоящей работе, с экспериментальными данными. В работе [21] была определена энергия ГЗ в Ni и получено значение ~0.9 Дж/м2, что близко к среднему значению энергии ГЗ, полученному в настоящей работе. Это позволяет утверждать, что значения энергий ГЗ, рассчитанные в настоящей работе, достаточно надежны.
В табл. 2 приведена оцененная ширина ГЗ, рассмотренных в настоящей работе. Видно, что почти для всех границ ширина составляет ~0.5 нм. Только для двойниковых ГЗ ширина границы заметно меньше. Эти результаты хорошо согласуются с данными диффузионных исследований, согласно которым диффузионная ширина границы зерна составляет 0.4–0.6 нм [22–24]. Близкие значения ширины ГЗ дают структурные исследования, выполненные с помощью электронной микроскопии высокого разрешения [25, 26].
В настоящей работе были определены энергии образования вакансий во всех рассмотренных ГЗ. В табл. 1 приведена минимальная энергия образования вакансий во всех рассмотренных границах. В литературе почти нет данных об энергиях образования вакансий в границах, рассматриваемых в данной работе. Лишь в работе [20] приведены результаты первопринципных расчетов энергии образования вакансий в границах $\Sigma 11,(113)\left[ {1\bar {1}0} \right]$ и $\Sigma 3,(111)\left[ {1\bar {1}0} \right].$ Приводимые в этой работе значения достаточно близки к полученным в настоящем исследовании.
Были проведены расчеты энергии образования вакансий в зависимости от расстояния от плоскости границы. Структура большинства ГЗ симметрична относительно плоскости границы. В этом случае зависимости энергии образования вакансий от расстояния от плоскости границы также симметричны. Это характерно, например, для границы $\Sigma 9,(221)\left[ {1\bar {1}0} \right],$ θ = 141.1°, как показано на рис. 4a. В то же время для некоторых границ наблюдается асимметрия структуры относительно плоскости границы.
Рис. 4.
Зависимости энергии образования вакансий от расстояния от границы для $\Sigma 9,\,\,(221)\left[ {1\bar {1}0} \right]$ (а) и $\Sigma 33,\,\,(118)\left[ {1\bar {1}0} \right]$ (б).
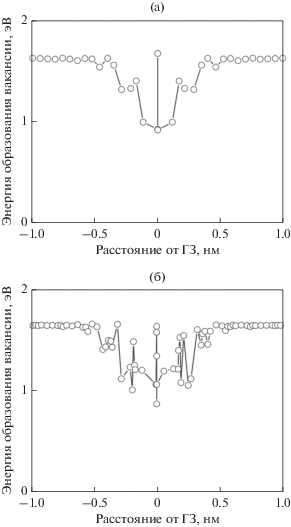
Это не всегда четко видно на изображениях структуры, но проявляется в зависимостях энергии образования вакансий от расстояния от границы. Это относится, например, к границе $\Sigma 33,(118)\left[ {1\bar {1}0} \right],$ θ = 20.1°, зависимость энергии образования вакансии от расстояния от границы для которой показана на рис. 4б. Аналогичный эффект наблюдался в работе [7] при моделировании ГЗ в W.
Ширина приграничной области, для которой энергия образования вакансий существенно меньше, чем энергия образования вакансий в объеме, составляет приблизительно 1 нм. Почти такое же значение приводится в работе [27]. Это приблизительно в два раза больше, чем структурная и диффузионная ширина ГЗ.
На рис. 5 показана исходная релаксированная структура границы $\Sigma 9,(221)\left[ {1\bar {1}0} \right]$ и ее структура после образования вакансий в соответствующих позициях и последующей релаксации.
Рис. 5.
Исходная релаксированная структура границы $\Sigma 9,(221)\left[ {1\bar {1}0} \right]$ (а) и ее структура после образования вакансий в позициях 1 (б), 2 (в) и 3 (г) и последующей релаксации. На рис. 5а цифрами показаны позиции образования вакансий. На рис. 5б–5г исходная позиция вакансии показана белым кружком.
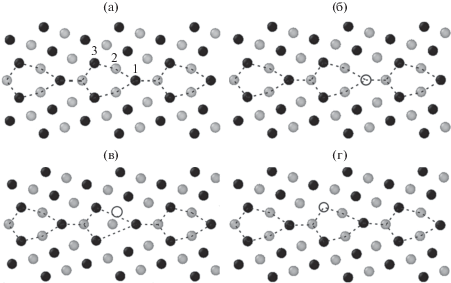
Энергии образования вакансий в позициях 1, 2 и 3 такой границы составляют, соответственно, 1.67, 0.99 и 1.40 эВ. Самое высокое значение энергии образования вакансии наблюдается для позиции 1, и оно близко к энергии образования вакансии в регулярной решетке. На рис. 5б представлена релаксированная структура ГЗ для этого случая. Видно, что после образования вакансии и последующей релаксации изменение позиций атомов в окрестности границы незначительно, то есть структура границы практически не меняется по сравнению с исходной.
В позиции 2 энергия образования вакансии минимальна (0.99 эВ). При этом изменения позиций атомов очень существенны и структура ГЗ в окрестности вакансии после релаксации значительно меняется (рис. 5в).
Промежуточное значение энергии образования вакансии (1.40 эВ) наблюдается для позиции 3. В этом случае после релаксации происходит заметный сдвиг ближайших к вакансии атомов, но изменения структуры ГЗ практически отсутствуют (рис. 5г).
Таким образом, чем более значительна релаксация структуры, сопровождающая образование вакансии, тем меньше энергия ее образования. Аналогичный эффект наблюдался в работе [7] при моделировании образования вакансий в границах зерен вольфрама.
На рис. 5б–5г исходная позиция вакансии показана белым кружком.
Следует также отметить, что вблизи ГЗ всегда есть позиции, для которых энергия образования вакансий заметно ниже, чем в объеме. Это заключение согласуется с результатами других исследований [7, 27–29].
ЗАКЛЮЧЕНИЕ
Выполнено компьютерное моделирование структуры и энергии симметричных границ наклона с осью 〈110〉 в поликристаллическом Ni. С помощью молекулярно-динамического моделирования проверена устойчивость структуры границ зерен при нагреве. Показано, что структуру симметричных границ наклона 〈110〉 можно представить ограниченным числом структурных элементов. Рассчитаны значения энергии и ширины границ, имеющих различные разориентировки. Установлено, что рассчитанная ширина границ близка к значениям, полученным в диффузионных экспериментах и электронно-микроскопических исследованиях высокого разрешения. Определены энергии образования вакансий в окрестности границ зерен.
Продемонстрирована разная степень релаксации структуры в ГЗ в зависимости от энергии образования вакансии, на примере границы $\Sigma 9,(221)\left[ {1\bar {1}0} \right].$
Список литературы
Sutton A.P., Balluffi R.W. Interfaces in Crystalline Materials. Clarendon Press, Oxford, 1995. 819 p.
Van Swygenhoven H., Farkas D., Caro A. Grain-boundary structures in polycrystalline metals at the nanoscale // Phys. Rev. 2000. V. 62. № 2. P. 831–838.
Movahedi-Rad A., Alizadeh R. Simulating Grain Boundary Energy Using Molecular Dynamics // J. Modern Physics. 2014. V. 5. P. 627–632.
Rittner J.D., Seidman D.N. 〈110〉 symmetric tilt grain-boundary structures in fcc metals with low stacking-fault energies // Phys. Rev. B. 1996. V. 54. № 10. P. 6999–7015.
Foiles S.M., Baskes M.I., Daw M.S. Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys // Phys. Rev. B. 1986. V. 33. № 5. P. 7983.
Plimton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics // J. Comp Phys. 1995. V. 117. P. 1–19.
Ступак М.Е., Уразалиев М.Г., Попов В.В. Структура и энергия симметричных границ наклона 〈110〉 в поликристаллическом W // ФММ. 2020. Т. 121. № 8. С. 877–883.
Tschopp M.A., McDowell D.L. Structures and energies of Sigma 3 asymmetric tilt grain boundaries in Cu and Al // Phil. Mag. 2007. V. 87. P. 3147–3173.
Mishin Y., Farkas D., Mehl M.J., Papaconstantopoulos D.A. Interatomic potentials for monoatomic metals from experimental data and ab initio calculations // Phys. Rev. B. 1999. V. 59. № 5. P. 3393–3407.
Zhou X.W., Johnson R.A., Wadley H.N.G. Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers // Phys. Rev. B. 2004. V. 69. № 14. P. 144113.
Stoller R.E., Tamm A., Béland L.K., Samolyuk G.D., Stocks G.M., Caro A., Slipchenko L.V., Osetsky Y.N., Aabloo A., Klintenberg M., Wang Y. Impact of Short-Range Forces on Defect Production from High-Energy Collisions // J. Chem. Theory and Computation. 2016. V. 12. № 6. P. 2871–2879.
Mendelev M.I., Kramer M.J., Hao S.G., Ho K.M., Wang C.Z. Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy // Phil. Mag. 2012. V. 92. № 35. P. 4454–4469.
Горелик С.С., Скаков Ю.А., Расторгуев Л.Н. Рентгенографический и электронно-оптический анализ: Учебн. пособие для вузов. 4-е изд. М.: “МИСИС”, 2002. 360 с.
Simons G., Wang H. Single Crystal Elastic Constants and Calculated Aggregate Properties. MIT Press, Cambridge, MA. 1977. 370 p.
Metal Reference Book, 5th ed / edited by C. J. Smith. Butterworth, London. 1976. 1552 p.
Wycisk W., Feller-Kniepmeier M.J. Quenching experiments in high purity Ni // Nucl. Mater. 1978. V. 69. № 1–2. P. 616–619.
Murr L.E. Interfacial Phenomena in Metals and Alloys. Addison-Wesley, Advanced Book ProgramReading. 1975. 376 p.
Stukowski A. Visualization and analysis of atomistic simulation data with OVITO – the Open Visualization Tool Modelling Simul // Mater. Sci. Eng. 2010. V. 18. P. 015012.
Takata N., Ikeda K., Yoshida F., Nakashima H., Abe H. Grain Boundary Structure and its Energy of 〈110〉 Symmetric Tilt Boundary in Copper // Mater. Sci. Forum. 2004. V. 467–470. P. 807–812.
Chen J., Dongare A.M. Role of grain boundary character on oxygen and hydrogen segregation-induced embrittlement in polycrystalline Ni // J. Mater. Sci. 2017. V. 52. P. 30–45.
Tyson W.R., Miller, W.A. Surface free energies of solid metals: Estimation from liquid surface tension measurements // Surface Sci. 1977. V. 62. № 1. P. 267–276.
Surholt T., Mishin Yu., Herzig Chr. Grain-boundary diffusion and segregation of gold in copper: Investigation in type-B and type-C kinetic regimes // Phys. Rev. B. 1994. V. 50. № 6. P. 3577–3587.
Divinski V.S., Reglitz G., Wilde G. Grain boundary self-diffusion in polycrystalline nickel of different purity levels // Acta Materi. 2010. V. 58. P. 386–395.
Prokoshkina D., Esin V.A., Wilde G., Divinski S.V. Grain boundary width, energy and self-diffusion in nickel: effect of material purity // Acta Mater. 2013. V. 61. № 14. P. 5188–5197.
Thomas G.J., Siegel R.W., Eastman J.A. Grain boundaries in nanophase palladium: High resolution electron microscopy and image simulation // Scr. Metall. Mater. 1990. V. 24. P. 201–206.
Fultz B., Kuwano H., Ouyang H. Average widths of grain boundaries in nanophase alloys synthesized by mechanical attrition // J. Appl. Phys. 1995. V. 77. P. 3458–3466.
Hallil A., Metsu A., Bouhattate J., Feaugas X. Correlation between vacancy formation and Σ3 grain boundary structures in nickel from atomistic simulations // Phil. Mag. 2016. V. 96. № 20. P. 2088–2114.
Suzuki A., Mishin Y. Atomistic Modeling of Point Defects and Diffusion in Copper Grain Boundaries // Interface Sci. 2003. V. 11. P. 131–148.
Chen N, Niu L.-L., Zhang Y., Shu X., Zhou H.-B., Jin S., Ran G., Lu G.-H., Gao F. Energetics of vacancy segregation to [100] symmetric tilt grain boundaries in bcc tungsten //Scientific Reports. 2016. V. 6. P. 36955.
Дополнительные материалы отсутствуют.
Инструменты
Физика металлов и металловедение