

Физиология растений, 2021, T. 68, № 4, стр. 371-379
JetGene – интернет-ресурс для анализа регуляторных областей или нуклеотидных контекстов у дифференциально транслируемых транскриптов растений
Н. С. Садовская a, *, О. Н. Мустафаев b, c, А. А. Тюрин a, И. В. Дейнеко a, И. В. Голденкова-Павлова a
a Институт физиологии растений им. К.А. Тимирязева Российской академии наук
Москва, Россия
b Бакинский государственный университет
Баку, Азербайджан
c Институт генетических ресурсов Национальной академии наук Азербайджана
Баку, Азербайджан
* E-mail: nataliya.sadovskaya@gmail.com
Поступила в редакцию 02.12.2020
После доработки 20.12.2020
Принята к публикации 20.12.2020
Аннотация
Различные регуляторные коды, содержащиеся в мРНК, могут определять судьбу любого транскрипта в процессе трансляции. Для поиска подобных регуляторных кодов и изучения их влияния на эффективность трансляции, мы разработали интернет-ресурс JetGene (https://jetgene.bioset.org/). Он содержит CDS, cDNA, 5'-UTR, 3'-UTR последовательности из шести основных групп живых организмов, включая растения. Данный интернет-ресурс имеет дружественный интерфейс, соединяет воедино широкий набор опций, предназначенный для сравнительного анализа нуклеотидных последовательностей и позволяет (1) оценить вариации длины, нуклеотидного состава, частоты использования кодонов, проанализировать GC-состав, CpG-острова, окружение стартового кодона и др.; (2) выявить и определить статистически значимую представленность потенциальных регуляторных контекстов в мРНК с разной эффективностью трансляции. Пользователь может сделать разносторонний in silico анализ полноразмерных или усеченных транскриптов, а также их кодирующих/некодирующих областей. Каждый этап анализа сопровождается графической интерпретацией результатов.
ВВЕДЕНИЕ
Трансляция представляет собой фундаментальный процесс и важную начальную точку в регуляции экспрессии генов для клеток всех живых организмов, поскольку в этом процессе раскрывается кодирующий потенциал мРНК через молекулу белка. В настоящее время превалирующее мнение о трансляционном контроле состоит в том, что он играет ключевую роль в совокупности клеточных процессов растений, например, в их ответе на всевозможные факторы окружающей среды и различные метаболиты [1]. Особого внимания заслуживает выявленная несоразмерность уровней мРНК с их белковыми продуктами, которая характерна для клеток эукариот в целом и для клеток растений в частности [2]. Континуум различных и, в тоже время, изящных экспериментальных исследований указывает на то, что в процессе декодирования генома, наряду с каноническими трансляционными правилами, довольно часто могут применяться правила регуляции и декодирования более высокого уровня. Это свидетельствует о наличии специфических регуляторных кодов, участвующих в трансляции мРНК растений.
Следует напомнить, что мРНК включают в себя 5'-нетранслируемую область (5'-UTR), кодирующий регион (CDS) и 3'-нетранслируемую область (3'-UTR), которые модулируют трансляцию в ряде “контрольных точек”: инициации, элонгации и терминации трансляции. Согласно текущему мнению, многочисленные регуляторные коды могут быть скрыты в нуклеотидных контекстах этих областей мРНК. Каждый из этих кодов в отдельности или несколько из них в сочетании друг с другом могут определять дальнейшую судьбу любого транскрипта в процессе трансляции [2]. Для выявления подобных регуляторных кодов применяют in silico анализ вышеперечисленных областей мРНК – CDS, 5'-UTR и 3'-UTR.
С целью обнаружения таких регуляторных кодов в мРНК и их корреляции с трансляционной эффективностью мы создали интернет-ресурс JetGene (https://jetgene.bioset.org/). Кроме того, JetGene позволяет оценить вариации нуклеотидного состава, частоты использования кодонов, оценить окружение стартового кодона и многое другое.
МАТЕРИАЛЫ И МЕТОДЫ
Мотивация создания JetGene. При создании JetGene задача состояла в том, чтобы предоставить пользователям, имеющим минимальный опыт в области биоинформатики и/или в программировании, простой и в тоже время удобный инструментарий для анализа и планирования эксперимента. Таким образом, в JetGene собран воедино широкий набор опций, предназначенный для сравнительного анализа последовательностей, который дает возможность (1) оценить вариации длины, нуклеотидного состава, частот использования кодонов, окружение стартового кодона трансляции; (2) выявить и определить статистически значимую представленность потенциальных регуляторных контекстов у мРНК с разной эффективностью трансляции.
Следует отметить, что JetGene содержит последовательности CDS, cDNA, 5'-UTR, 3'-UTR для шести царств живых организмов, включая 45 видов растений. Удобный функциональный набор инструментов JetGene позволяет проводить исследование как по полноразмерным транскриптам, так и по усеченным транскриптам, а также по кодирующим/некодирующим областям мРНК.
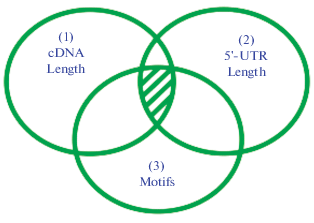
Алгоритм “Система вложенных выборок”. Еще одно важное преимущество JetGene заключается в реализации в нем особого алгоритма, названного нами “Система вложенных выборок”. Его суть состоит в том, что на первом этапе работы исследователь выбирает определенный критерий в качестве основного, например, (1) cDNA заданной длины, “cDNA Length”, и формирует главную выборку. На дальнейших этапах анализа он может использовать оставшиеся параметры как дополнительные, например, (2) добавить вспомогательный критерий “5'-UTR Length”, что позволит отобрать из главной выборки последовательности, имеющие определенную длину 5'-UTR, и сформировать выборку последовательностей второго порядка; (3) затем можно добавить следующий параметр, например, поиск по мотиву “Motifs”, в результате которого JetGene выберет последовательности, содержащие указанный мотив из выборки второго порядка. Таким образом, имеется возможность сформировать серию последующих выборок, каждая из которых строится на основе предыдущей без извлечения промежуточных результатов из JetGene (рис. 1). Старшинство критериев (основной и вспомогательные) пользователь может определить по своему усмотрению. В результате исследователь имеет возможность получить различные варианты биологических текстов, удовлетворяющие нетривиальным сочетаниям параметров, при этом количество таких комбинаций не ограничено. Следует подчеркнуть, что как итоговая, так и каждая из промежуточных выборок могут быть экстрагированы из JetGene в fasta-формате на произвольном этапе работы.
Рис. 1.
Алгоритм “Система вложенных выборок”. Окружности схематически изображают возможность выбрать последовательности по критериям “cDNA Length”, “5'-UTR Length”, “Motifs”. В качестве начального критерия выбран (1) размер cDNA, в качестве сопровождающих – (2) размер 5'-UTR и (3) GC-состав выше определенного значения. Результирующая выборка пользователя (4) находится на пересечении всех окружностей и заштрихована.
Строение JetGene. Транскриптомные данные о представителях шести ключевых царств живых организмов, включая растения, загружены с сервера Ensembl https://www.ensembl.org/ [3] 28 июня 2017 г. и регулярно обновляются в JetGene (раз в неделю). Описание каждого транскриптома содержит информацию о сборке и количестве находящихся в нем последовательностей. Для большинства эукариот главный интерфейс JetGene состоит из четырех основных разделов (“CDS data”, “cDNA data”, “5'-UTR data”, “3'-UTR data”) (рис. 2). Следует отметить, что информация об UTR получена путем вычитания CDS из cDNA. Для ряда организмов приведена генная онтология (Gene Ontology Annotations, GO) [4]. Раздел GO является вспомогательным. Он не связан с основными разделами и присутствует только в том случае, когда информация о генной онтологии приведена на сервере Ensembl.
Рис. 2.
Общая структура JetGene. Стрелки символизируют, что работу можно начать с любого раздела (CDS, cDNA, 5'‑URT, 3'-UTR) и переходить в любой раздел, не извлекая полученный результат из JetGene (a). Схематическое изображение гена на хромосоме (интроны удалены) (б).

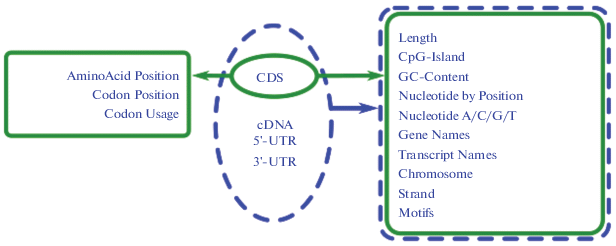
JetGene организован в модульной форме. Модули можно использовать как индивидуально, так и применять их последовательно с целью проведения расширенного и непрерывного исследования. Веб-интерфейс содержит 10 основных модулей, свойственных четырем основным разделам, и 3 приложения, присущие разделу “CDS data”. Список модулей, характерных для каждого раздела (“CDS data”, “cDNA data”, “5'-UTR data”, “3'-UTR data”), приведен на рис. 3.
Рис. 3.
Обзор основных модулей JetGene. Схематически изображены модули, доступные для каждого из разделов “CDS data”, “cDNA data”, “5'-UTR data”, “3'-UTR data”.
Кроме того, реализована возможность загрузить произвольную выборку нуклеотидных последовательностей и провести последующий анализ (эта опция доступна после бесплатной регистрации). В этом случае не будет показана разметка последовательностей cDNA на CDS, 5'-UTR, 3'-UTR, а последовательности будут интерпретированы JetGene как CDS. Исследователю будут доступны все приложения, за исключением “Chromosome” и “Strand”. Возможность экстрагировать полученные последовательности в fasta-формате на произвольной стадии исследования также сохраняется.
JetGene обладает дружественным интерфейсом. Кроме того, на каждом этапе работы в нем предусмотрена графическая интерпретация результатов проведенного исследования, которая сопровождается статистически обработанными количественными значениями. Это дает возможность сравнить выборку пользователя, созданную в процессе работы, с информацией по всему транскриптому изучаемого организма, и тем самым наглядно проиллюстрировать полученные результаты анализа. Все вышесказанное позволяет выполнить комплексный in silico анализ для огромного количества научных направлений. Таким образом, JetGene можно выбрать в качестве отправной точки научных исследований и удобного интернет-ресурса для широкого спектра работ.
Далее мы даем список модулей, характерных для каждого их четырех основных типов данных (“CDS data”, “cDNA data”, “5'-UTR data”, “3'-UTR data”).
Модули, специфичные только для “CDS data”:
1. AminoAcid Position;
2. Codon Position;
3. Codon Usage.
Модули, специфичные для “CDS data”, “cDNA data”, “5'-UTR data”, “3'-UTR data”:
1. CDS/cDNA/5'-UTR/3'-UTR Length;
2. CpG-Island in CDS/cDNA/5'-UTR/3'-UTR;
3. GC-Content in CDS/cDNA/5'-UTR/3'-UTR;
4. Nucleotide by Position in CDS/cDNA/5'-UTR/3'-UTR;
5. Nucleotide A/C/G/T in CDS/cDNA/5'-UTR/3'-UTR;
6. Gene Names;
7. Transcript Names;
8. Chromosome;
9. Strand;
10. Motifs.
РЕЗУЛЬТАТЫ
Ниже приведено краткое описание модулей, свойственных для каждого из четырех основных разделов JetGene. В таблице (Дополнительные материалы) представлены потенциальные области использования модулей JetGene.
Модули, характерные для “CDS data”
AminoAcid Position. В текущей опции реализована возможность отображать аминокислоту, локализованную в позиции 1–10 как с C-, так и с N‑концевой области CDS. Она может быть полезна для применения правила “N-конца”, согласно которому время полужизни белка определяется вторым N-концевым аминокислотным остатком его полипептидной цепи, в частности, для дизайна 5’-концевой области целевого гена либо для анализа и конструирования сигнальных пептидов (Дополнительные материалы таблица).
Codon Position. Данная утилита аналогична “AminoAcid Position”. Она определяет, какие триплеты расположены в позиции 1–10 с 5'- или с 3'‑концевой области CDS. И позволяет, например, исследовать N-концевую область белка или сигнальных пептидов на нуклеотидном уровне.
Codon Usage. Текущий модуль показывает триплеты, кодирующие аминокислоты в CDS, а также их численный и процентный состав (за 100% принимается сумма всех триплетов, кодирующих данную аминокислоту, а не сумма всех кодонов по CDS). Он позволяет исследовать полноразмерные CDS или их усеченный вариант (опция “Sequence region to calculate data (%)”). Кроме того, в этом модуле предусмотрена возможность загрузить и проанализировать любую нуклеотидную последовательность. Подобная утилита может оказаться полезна в случае, когда экспериментатору необходимо сконструировать целевую последовательность таким образом, чтобы ее кодонный состав был схож с кодонным составом генома хозяина, что, в свою очередь, приводит к увеличению уровня трансляции целевого белка (Дополнительные материалы, таблица).
Модули, характерные для “CDS data”, “cDNA data”, “5'-UTR data”, “3'-UTR data”
CDS/cDNA/5'-UTR/3'-UTR Length. Эта опция дает распределение всех последовательностей CDS/cDNA/5'-UTR/3'-UTR по длинам в изучаемом организме. Кроме этого, имеется возможность выбрать последовательности определенной длины (разбивка ведется с шагом по 500 н.), либо задать диапазон, интересующий пользователя, на линейке “Values interval to calculate data”. Данное приложение может быть использовано при подборе целевых генов максимальной длины, которые можно клонировать в выбранный вектор, или в исследованиях по оценке взаимосвязи между длиной 5'-UTR и эффективностью трансляции мРНК (Дополнительные материалы, таблица). Например, в исследовании взаимосвязи между длиной 5'-UTR и нагрузкой транскриптов рибосомами у Arabidopsis thaliana было продемонстрировано, что (1) длина 5'-UTR 50–75 н. обеспечивает оптимальную нагрузку рибосомами, а (2) короткие 5'-UTR (менее 25 н.) и длинные 5'-UTR (175–300 н.) могут ингибировать нагрузку транскрипта рибосом, т.е. установлена общая закономерность влияния длины 5'-UTR на трансляционный статус мРНК (Дополнительные материалы, таблица).
CpG-Island in CDS/cDNA/5'-UTR/3'-UTR. Данная утилита позволяет исследовать CpG-острова и вычислить процент CpG в CDS/cDNA/5'-UTR/3'-UTR. Она анализирует как полноразмерные последовательности, так и их усеченные варианты (опция “Sequence region to calculate data (%)”). Этот модуль дает возможность отобрать все последовательности, для которых процент CpG-Island попадает в интервал, заданный пользователем (опция “Values interval to calculate data”). Такой анализ может быть полезен для изучения различий в метилировании CpG-богатых последовательностей из разных организмов, в том числе растений (Дополнительные материалы, таблица).
GC-Content in CDS/cDNA/5'-UTR/3'-UTR. Описываемая утилита аналогична “GpC-Island in CDS/cDNA/5'-UTR/3'-UTR”, но учитывает все G и C нуклеотиды, входящие в состав транскриптов. Имеется возможность выбрать все транскрипты, обладающие определенным GC-составом (разбивка ведется с шагом 1%). Она может быть применена в исследованиях по оценке распределения содержания динуклеотидов GC в кодирующих последовательностях, что может сказываться на эффективности экспрессии генов с различным соотношением GC динуклеотидов (Дополнительные материалы, таблица).
Nucleotide by Position in CDS/cDNA/5'-UTR/3'-UTR. Указанный модуль показывает, какой нуклеотид расположен в позиции 1–10 как с 5'- так и с 3'-концевой области CDS/cDNA/5'-UTR/3'-UTR. Он может быть полезен в сравнительных исследованиях полных геномов растений для выявления взаимосвязи между высококонсервативными позициями (преобладанием повторов трех нуклеотидов вокруг старта инициации трансляции – TIS) и уровнем белка (Дополнительные материалы, таблица). Например, такой анализ позволил установить самые сильные детерминанты эффективности трансляции у растений, расположенные в непосредственной близости от стартового кодона: A/G–3 и G+4 [или A/G–3N–2N–1AUG и AUGG+4]. В сходных исследованиях убедительно продемонстрировано, что С+5 важен для эффективности трансляции, а на примере различных генов A. thaliana выяснено, что область от позиции –5 до –1 является наиболее важной для эффективности трансляции. При этом нуклеотиды A наиболее благоприятны, а нуклеотиды T неблагоприятны для трансляции, когда они локализованы в позициях от –4 до –1 в 5'-UTR (Дополнительные материалы таблица).
Nucleotide A/C/G/T in CDS/cDNA/5'-UTR/3'-UTR. Текущая опция определяет процентное содержание нуклеотида A/C/G/T в CDS/cDNA/5'-UTR/3'-UTR. Она анализирует как полноразмерные последовательности, так и их усеченные варианты (опция “Sequence region to calculate data (%)”). Имеется возможность выбрать все последовательности, для которых процент нуклеотида A/C/G/T попадает в интервал, заданный пользователем (опция “Values interval to calculate data”). Данная утилита позволяет сформировать для анализа выборки последовательностей, характеризующихся определенным нуклеотидным составом. Данное приложение может быть использовано в исследованиях, в которых необходимо установить взаимосвязь между композицией моно- и динуклеотидов в 5'-UTR и нагрузкой транскриптов рибосомами у растений (Дополнительные материалы таблица). В частности, для A. thaliana показано, что: (1) мРНК с высокой рибосомной нагрузкой обычно имели 5'-UTR с высоким содержанием аденина (A) и динуклеотидов AU и AC; (2) плохо нагруженные рибосомами мРНК обычно имели 5'-UTR с повышенным содержанием гуанина (G), и динуклеотида GU (Дополнительные материалы, таблица).
Gene Names. Этот модуль позволяет выбрать все последовательности, имеющие имя или общую часть имени. Дополнительно имеется возможность загружать выборку пользователя по именам генов, если данные по исследуемому организму охвачены JetGene. Следует отметить, что в этом случае пользователю будут доступны все разделы JetGene.
Transcript Names. Текущее приложение аналогично “Gene Names”, но при этом пользователь может найти как уникальный транскрипт(ы), так и все транскрипты, относящиеся к одному гену или имеющие общую часть имени гена, введенного пользователем в строку поиска. Модуль позволяет легко выявить все изоформы изучаемого гена и проанализировать наличие различий между ними.
Chromosome. Указанная опция показывает распределение последовательностей по хромосомам, а также на митохондриальной ДНК. Она может быть полезна в случае, когда пользователя интересуют последовательности, локализованные на определенной хромосоме, либо при сравнении данных, полученных для двух разных хромосом.
Strand. Эта подпрограмма дает возможность выяснить, какие CDS локализованы на прямой, а какие – на обратной цепи ДНК, и быстро разделить всю выборку на две соответствующие части. Для бактерий такая нехитрая процедура позволяет выявить ошибочное предписание генов к одному оперону. Кроме того, она может быть полезна в работах по оценке длины генов в зависимости от их расположения на лидирующей (прямой) или на запаздывающей (обратной) цепях (Дополнительные материалы таблица).
Motifs. Указанный модуль позволяет провести поиск последовательностей, содержащих определенные мотивы, в том числе одновременно несколько мотивов, указанных пользователем (оператор AND), или хотя бы один из них (оператор OR). Анализ можно проводить как по полноразмерным последовательностям, так и по области, ограниченной пользователем. Результаты выводятся в виде гистограммы, отображающей частоту встречаемости мотива по блокам, на которые разбиваются последовательности. Для каждого введенного мотива исследователь имеет возможность указать определенные блоки в таблице. В этом случае выборка будет ограничена теми генами, в которых мотив встречается в указанных регионах. Описанный модуль может быть полезен при выявлении мотивов внутри регуляторных последовательностей, определении их функционального значения в обеспечении уровня экспрессии целевого гена(-ов) и при конструировании синтетических регуляторных последовательностей (Дополнительные материалы таблица). Как, например, в исследовании 5'-UTRs из 15 971 гена A. thaliana, в котором выявлены мотивы TAGGGTTT и AAAACCCT, характерные для многих генов. Это потенциально указывает на их вклад в эффективность трансляции. Транскриптомное сравнение мРНК, не связанных с полисомами, и полисомных мРНК убедительно показало наличие светового запуска трансляционного контроля для транскриптов, имеющих один из этих двух мотивов в 5'-UTR, но не для транскриптов, представленных на высоком уровне. Данное наблюдение позволило предположить, что трансляционный контроль за счет элементов, перечисленных выше, может обеспечивать дифференциальную трансляцию мРНК. Дополнительно авторы продемонстрировали, что мотивы TAGGGTTT и AAAACCCT, представляющие собой цис-элементы, могут играть роль в трансляционном управлении, в то время как A. thaliana реорганизует протеом в ответ на внешние экологические стимулы. Дальнейшие исследования экспериментально доказали, что мотив TAGGGTTT регулирует экспрессию гена именно на трансляционном уровне. Необходимо отметить, что комплементарность последовательностей этих двух элементов позволила предположить, что они могут функционировать в процессе контроля трансляции за счет формирования шпилеподобной структуры на мРНК. Однако, изучение появления этих мотивов в 5'-UTR транскриптов, кодирующих строго подтвержденные белковые продукты, позволило выявить только 4 гена, содержащие оба этих мотива AAAACCCT и TAGGGTTT. Таким образом, структура спаривания оснований, образованная этими двумя цис-элементами, не может объяснить трансляционную регуляцию большинства транскриптов. Кроме того, доказано, что эти два элемента могут независимо участвовать в контроле трансляции.
ОБСУЖДЕНИЕ
Сравнение с другими базами данных. Мы разработали интернет-ресурс JetGene, который очень прост в использовании и ориентирован не только на опытных биоинформатиков, но и на экспериментаторов, имеющих ограниченные знания в in silico анализе и в программировании. Сравним его с другими веб-ресурсами.
В настоящее время биологические тексты последовательностей хранятся на серверах различных источников. Наиболее часто в них представлена CDS и соответствующая ей аминокислотная последовательность, как, например, в GenBank [5] или в KEGG [6]. В отличие от JetGene, они содержат различные вспомогательные опции, такие как карты метаболических путей, пакет программ Blast [7] для поиска гомологичных последовательностей, список публикаций, ссылки на внешние интернет-ресурсы, дающие всестороннее описание изучаемого гена или белка, и многое другое. Несмотря на разносторонность представленной информации, при работе с подобными онлайн-ресурсами выбор последовательностей возможен только на самом тривиальном уровне: найти последовательность с заданной функцией или гомологичную данной.
Далее следует остановиться на ресурсах, позволяющих проводить комплексный поиск последовательностей. К ним относится Ensembl https:// www.ensembl.org/ [3], который послужил основой для JetGene. Следует отметить, что вся информация о нуклеотидных последовательностях, представленных в Ensembl, содержатся в JetGene. В настоящее время Ensembl представляет собой один из важнейших интернет-ресурсов, в котором хранится информация об аннотации генов, генетике, сравнительной геномике и эпигеномике для колоссального количества живых организмов. Для многих из них представлен не один, а несколько вариантов сборки генома. Возможности использования Ensembl варьируются от быстрого просмотра данных до биоинформатического анализа в масштабах всего генома. При этом, чтобы обеспечить доступ к сведениям, интересующим пользователя, Ensembl предлагает доступ через BioMart [8], через различные языки программирования и REST APIs [9, 10] или через FTP. Однако BioMart не в полной мере использует информацию, хранящуюся в Ensembl (например, отображает малую часть организмов, информация о которых содержится в Ensembl), а использование REST APIs и FTP требует навыков программирования, которыми обладают не все заинтересованные пользователи.
BioMart дает возможность работать отдельно с CDS, cDNA, 5'-UTR, 3'-UTR, а также с белковыми последовательностями. Для поиска нужной информации пользователь может составить набор фильтров таким образом, чтобы охватить широкий диапазон данных, начиная от положения последовательности на хромосоме и заканчивая фенотипом организма. Общая информация, хранящаяся в BioMart, существенно превосходит тот объем информации, который содержится в JetGene. Набор опций BioMart также заметно превышает набор модулей JetGene. В частности, BioMart позволяет задать координаты на хромосоме, получить информацию об интрон-экзонной структуре, найти ортологи в других организмах и многое другое. При этом пересечение опцией между BioMart и JetGene незначительное. В частности, BioMart, как и JetGene, позволяет выбрать хромосому для анализа, осуществить поиск по “gene ID”, дает возможность отобразить последовательности CDS, cDNA, 5'-UTR, 3'-UTR и получить их в fasta-формате, сделать поиск по GO. При этом, столь важная для исследователя информация, как, например, длина последовательности, ее GC-состав, локализация изучаемой последовательности на прямой/обратной цепи и т.д., отображается исключительно в результирующем файле. Дальнейший отбор последовательностей по каждому из вышеперечисленных параметров необходимо делать вручную. Следует упомянуть, что часть информации, например, процентное содержание нуклеотида A/C/G/T или какой нуклеотид расположен в позиции 1–10, распределение триплетов внутри исследуемой выборки последовательностей, не предоставлена в BioMart ни в виде опций, ни в результирующем файле. Кроме того, возможность работать с усеченными последовательностями представлена в BioMart не столь наглядно как в JetGene, а графическая интерпретация результатов по выбранному параметру отсутствует. Также существует ряд ограничений при попытке сделать несколько итераций работы или при попытке осуществить переход CDS/cDNA/5'-UTR/3'-UTR. Т.е., например, затруднительно перейти напрямую от анализа 5'-UTR к анализу cDNA, в которых они содержатся, без дополнительных вспомогательных действий.
UCSC Genome Browser https://genome.ucsc.edu/ [11], представляет собой еще один веб-ресурс, позволяющий сделать комплексный поиск и анализ последовательностей. Он содержит данные более чем для 100 видов, причем для ряда из них предложено несколько вариантов сборки. При этом UCSC Genome Browser охватывает меньше царств, чем JetGene, а в каждом из них содержится меньше организмов, чем в JetGene. Например, в нем отсутствуют сведения о растениях, а информация о грибах приведена только для Saccharomyces cerevisiae. Для отбора последовательностей по критериям, интересующим пользователя, и для получения выборки в fasta-формате, исследователь может использовать опцию UCSC Table Browser, который, подобно JetGene, позволяет составить выборку последовательностей по нескольким параметрам и экстрагировать ее в fastа-формате. Тем не менее, настройки Table Browser менее наглядны, чем настройки JetGene. Для того чтобы суметь сформировать корректный запрос, загрузить свои данные или использовать информацию, представленную в этом интернет-ресурсе, а также применить несколько критериев запроса и составить пересечение/объединение данных между собой, исследователь должен обладать определенными биоинформатическими знаниями. Ему необходимо изучить структуру формата входных/ выходных данных и описание фильтров. Более того, пользователю требуется подготовить материал, на основе которого будет строиться пересечение критериев. При регулярном решении одинаковых задач это оправдано. Тем не менее, изучение настроек занимает значительное время при быстро меняющихся задачах, а также при подборе последовательностей по широкому спектру критериев. Следует отметить, что графическая интерпретация в UCSC Table Browser не представлена.
В то же время данные из UCSC Table Browser можно экспортировать непосредственно в открытую веб-платформу Galaxy http://usegalaxy.org [12]. Это занимает дополнительное время. При этом ряд ее опций совпадает с утилитами JetGene (например, анализ CDS, cDNA, 5'-UTR, 3'-UTR, анализ GC-состава, возможность выбрать последовательности определенной длины, возможность исследовать как полноразмерные последовательности, так и определенные участки целевых последовательностей, получить последовательности в fasta-формате и т.д.), но прописаны они не столь наглядно, как в JetGene. Также стоит отметить, что, как в Galaxy, так и в JetGene, нет ограничений на осуществление перехода между CDS/cDNA/5'-UTR/3'-UTR в процессе анализа. Но в Galaxy подобного рода переходы осуществляются менее тривиально и занимают больше времени, чем в JetGene.
Использование JetGene. Успешное применение JetGene (ранее база данных назвалась FlowGene) продемонстрировано в нескольких наших исследованиях. Так, в работе [13] изучен уровень транскрипции генов у растений в зависимости от нуклеотидного состава 5'-UTR. В ней применен алгоритм “Система вложенных выборок”. Основным критерием был выбран размер последовательностей не менее 200 н. (минимальный размер CpG-островка) (1). В качестве дополнительных критериев приняли: (2) GC-содержание выше 50% (одна из характеристик CpG-островков); (3) нуклеотиды, окружающие стартовый кодон, в положении –3 и +4, согласно последовательности Kozak; (4) отсутствие альтернативных стартовых и терминирующих кодонов. Далее в результирующей выборке искали 6-мерные мотивы, которые встречались не менее чем у 50% анализируемых последовательностей. Впоследствии эти мотивы были включены в дизайн синтетической последовательности и было получено экспериментальное подтверждение того, что 5'-области генов с высоким содержанием динуклеотидов CpG могут способствовать увеличению уровня транскрипции генов у растений. Еще в одном исследовании оптимизация кодонового состава gox гена Penicillium funiculosum с использованием ресурса JetGene позволила достичь эффективной экспрессии этого гена в растениях Solanum tuberosum, и как следствие обеспечить устойчивость к фитопатогену [14]. В другой работе [15] с применением алгоритмов ресурса JetGene проведен in silico анализ двух групп транскриптов с разной трансляционной эффективностью, что позволило установить, что пиримидиновые динуклеотиды и мотивы характерны для 5'-нетранслируемой области мРНК с высокой трансляционной эффективностью, тогда как пуриновые динуклеотиды и мотивы ассоциированы с транскриптами, имеющими низкую трансляционную эффективность.
Колебания нуклеотидного состава отмечены в геномах всех организмов, включая растения, и определяют эффективность экспрессии генов каждого вида [16, 17]. Знание точных механизмов регуляции является ключевым для понимания того, что заставляет организмы переключать гены. Использование информации о вариациях нуклеотидного состава важно при разработке противовирусных вакцин [18], дает возможность успешно оптимизировать последовательность целевого гена под кодоновый состав организма-хозяина, предсказать гены на основе геномных последовательностей [19], конструировать вырожденные праймеры [20] и многое другое. Изучение колебаний нуклеотидного состава занимает центральную позицию в таких важных областях как молекулярная эволюция [21] и биотехнология [22]. Наличие полногеномных последовательностей дает уникальную возможность определить закономерности в распределении различных свойств [23] как по всему геному, так и по областям отдельного транскрипта. Так, например, была выявлена связь между нуклеотидным составом и трансляцией белка [24, 25], уровнем экспрессии генов [26, 27], показано изменение нуклеотидного состава в зависимости от локализации последовательности в клетке [28], обнаружено влияние третьей позиции в кодоне в эффективность трансляции мРНК [29] и др.
Успех в проведении подобного рода исследований в значительной мере зависит от возможности сформировать наборы биологических текстов последовательностей, исходя из широкого ряда критериев, а также от возможности определить статистически значимые свойства в их распределении. Чем больше число параметров, участвующих в анализе, тем выше потенциал для создания и манипуляций выборками. Следовательно, тем шире потенциал для поиска и выявления характеристик, оказывающих влияние на биологические свойства последовательностей. В соответствии со всеми вышеперечисленными требованиями, нами разработан интернет-ресурс JetGene, который позволяет быстро и эффективно проводить анализ подобного рода. Следует отметить, что в настоящее время он дает наиболее полное представление о структурно-функциональном потенциале биологического текста. JetGene разработан для анализа исключительно нуклеотидных последовательностей и ориентирован на исследователей-экспериментаторов, не имеющих каких-либо специальных навыков в области in silico анализа или в программировании. Его уникальность заключается в том, что любой пользователь способен в кратчайшие сроки прозондировать огромные массивы информации, имеющиеся у него в распоряжении. Или составить de novo различные наборы последовательностей, удовлетворяющие задачам эксперимента, и быстро их проанализировать. Таким образом, исходя из критериев, интересующих пользователя, варьируя широким диапазоном параметров, можно провести полноценное биоинформационное исследование, составить выборку нуклеотидных последовательностей и извлечь ее из JetGene. Следует отметить, что графическая визуализация результатов сопровождает каждый шаг анализа и существенно облегчает работу пользователя.
Работа выполнена при поддержке гранта Российского научного фонда 18-14-00026.
Настоящая статья не содержит каких-либо исследований с участием людей и животных в качестве объектов. Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Goldenkova-Pavlova I.V., Pavlenko O.S., Mustafaev O.N., Deyineko I.V., Kabardaeva K.V. Tyurin A.A. Computational and experimental tools to monitor the changes in translation efficiency of plant mRNAs on a genome-wide scale: advantages, limitations, and solutions // Int. J. Mol. Sci. 2019. V. 20. P. 33. https://doi.org/10.3390/ijms20010033
Kabardaeva K.V., Tyurin A.A., Pavlenko O.S., Gra O.A., Deyneko I.V., Kouchoro F., Mustafaev O.N., Goldenkova-Pavlova I.V. Fine tuning of translation: a complex web of mechanisms and its relevance to plant functional genomics and biotechnology // Russ. J. Plant Physiol. 2019. V. 66. P. 835. https://doi.org/10.1134/s1021443719060074
Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R., Bhai J., Billis K., Boddu S., Marugán J.K., Cummins C. et al. Ensembl 2020 // Nucleic Acids Res. 2020. V. 48. P. D682. https://doi.org/10.1093/nar/gkz966
Carbon S., Douglass E., Dunn N., Good B., Harris N.L., Lewis S.E., Mungall C.J., Basu S., Chisholm R.L., Dodson R.J., Hartline E., Fey P., Thomas P.D., Albou L.P., Ebert D. et al. The Gene Ontology Resource: 20 years and still GOing strong // Nucleic Acids Res. 2019. V. 47. P. D330. https://doi.org/10.1093/nar/gky1055
Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank // Nucleic Acids Res. 2017. V. 45. P. D37. https://doi.org/10.1093/nar/gkw1070
Kanehisa M. KEGG bioinformatics resource for plant genomics and metabolomics // Methods Mol. Biol. 2016. V. 1374. P. 55. https://doi.org/10.1007/978-1-4939-3167-5_3
Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs // Nucleic Acids Res. 1997. V. 25. P. 3389. https://doi.org/10.1093/nar/25.17.3389
Kinsella R.J., Kähäri A., Haider S., Zamora J., Proctor G., Spudich G., Almeida-King J., Staines D., Derwent P., Kerhornou A., Kersey P., Flicek P. Ensembl BioMarts: a hub for data retrieval across taxonomic space // Database (Oxford). 2011. V. 2011. P. bar030 https://doi.org/10.1093/database/bar030
Yates A., Beal K., Keenan S., McLaren W., Pignatelli M., Ritchie G.R., Ruffier M., Taylor K., Vullo A., Flicek P. The Ensembl REST API: Ensembl data for any language // Bioinformatics. 2015. V. 31. P. 143. https://doi.org/10.1093/bioinformatics/btu613
Ruffier M., Kähäri A., Komorowska M., Keenan S., Laird M., Longden I., Proctor G., Searle S., Staines D., Taylor K., Vullo A., Yates A., Zerbino D., Flicek P. Ensembl core software resources: storage and programmatic access for DNA sequence and genome annotation // Database (Oxford). 2017. V. 2017. P. bax020. https://doi.org/10.1093/database/bax020
Hung J.H., Weng Z. Visualizing genomic annotations with the UCSC Genome Browser // Cold Spring Harb Protoc. V. 2016. P. 2016. https://doi.org/10.1101/pdb.prot093062
Goecks J., Nekrutenko A., Taylor J., Team G. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences // Genome Biol. 2010. V. 11. P. R86. https://doi.org/10.1186/gb-2010-11-8-r86
Tyurin A., Kabardaeva K., Gra O., Mustafaev O., Sadovskaya N., Pavlenko O., Goldenkova-Pavlova I. Efficient expression of a heterologous gene in plants depends on the nucleotide composition of mRNA’s 5'-region // Russ. J. Plant Physiol. 2016. V. 63. P. 511. https://doi.org/10.1134/s1021443716030158
Савчин Д.В., Вересова Т.Н., Межнина О.А., Панюш А.С., Вячеславова А.О., Голденкова-Павлова И.В. Оптимизация кодонового состава грибного гена gox Penicillium funiculosum для эффективной экспрессии в растениях Solanum tuberosum // Весцi НАН Беларусi. Сер. бiял. навук. 2015. № 1. С. 50.
Kabardaeva K.V., Turin A.A., Kouchoro F., Mustafaev O.N., Deineko I.V., Fadeev V.S., Goldenkova-Pavlova I.V. Regulatory contexts in the 5'-region of mRNA from Arabidopsis thaliana plants and their role in translation efficiency // Russ. J. Plant Physiol. 2020. V. 67. P. 425. https://doi.org/10.1134/S1021443720030139
Quax T., Claassens N., Söll D., van der Oost J. Codon bias as a means to fine-tune gene expression // Mol. Cell. 2015. V. 59. P. 149. https://doi.org/10.1016/j.molcel.2015.05.035
Song H., Gao H., Liu J., Tian P., Nan Z. Comprehensive analysis of correlations among codon usage bias, gene expression, and substitution rate in Arachis duranensis and Arachis ipaënsis orthologs // Sci. Rep. 2017. V. 7. P. 14853. https://doi.org/10.1038/s41598-017-13981-1
Lingemann M., Liu X., Surman S., Liang B., Herbert R., Hackenberg A., Buchholz U., Collins P., Munir S. Attenuated human parainfluenza virus type 1 expressing Ebola virus glycoprotein GP administered intranasally is immunogenic in African green monkeys // J. Virol. 2017. V. 91. P. e02469-16. https://doi.org/10.1128/JVI.02469-16
Picardi E., Pesole G. Computational methods for ab initio and comparative gene finding // Methods Mol. Biol. 2010. V. 609. P. 269. https://doi.org/10.1007/978-1-60327-241-4_16
Zhou T., Gu W., Ma J., Sun X., Lu Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses // Biosystems. 2005. V. 81. P. 77. https://doi.org/10.1016/j.biosystems.2005.03.002
Kim N., Lim S., Chae H., Park Y. Complete mitochondrial genome of the Amur hedgehog Erinaceus amurensis (Erinaceidae) and higher phylogeny of the family Erinaceidae // Genet. Mol. Res. 2017. V. 16. https://doi.org/10.4238/gmr16019300
Kinkema M., Geijskes J., Delucca P., Palupe A., Shand K., Coleman H., Brinin A., Williams B., Sainz M., Dale J. Improved molecular tools for sugar cane biotechnology // Plant Mol Biol. 2014. V. 84. P. 497. https://doi.org/10.1007/s11103-013-0147-8
Chaney J., Clark P. Roles for synonymous codon usage in protein biogenesis // Annu Rev Biophys. 2015. V. 44. P. 143. https://doi.org/10.1146/annurev-biophys-060414-034333
Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation // Cell. 2010. V. 141. P. 344. https://doi.org/10.1016/j.cell.2010.03.031
Saunders R., Deane Ch.M. Synonymous codon usage influences the local protein structure observed // Nucleic Acids Res. 2010. V. 38. P. 6719. https://doi.org/10.1093/nar/gkq495
Whittle C., Extavour C. Expression-linked patterns of codon usage, amino acid frequency, and protein length in the basally branching arthropod Parasteatoda tepidariorum // Genome Biol Evol. 2016. V. 8. P. 2722. https://doi.org/10.1093/gbe/evw068
Tian J., Yan Y., Yue Q., Liu X., Chu X., Wu N., Fan Y. Predicting synonymous codon usage and optimizing the heterologous gene for expression in E. coli // Sci. Rep. 2017. V. 7. P. 9926. https://doi.org/10.1038/s41598-017-10546-0
Diament A., Pinter R., Tuller T. Three-dimensional eukaryotic genomic organization is strongly correlated with codon usage expression and function // Nat. Commun. 2014. V. 5. P. 5876. https://doi.org/10.1038/ncomms6876
Shen W., Wang D., Ye B., Shi M., Ma L., Zhang Y., Zhao Zh. GC3-biased gene domains in mammalian genomes // Bioinformatics. 2015. V. 31. P. 3081. https://doi.org/10.1093/bioinformatics/btv329
Дополнительные материалы
- скачать ESM.docx
- Таблица 1. Потенциальные области использования модулей JetGene
СПИСОК ЛИТЕРАТУРЫ
Инструменты
Физиология растений