

Генетика, 2019, T. 55, № 10, стр. 1208-1217
Детекция точечных мутаций и хромосомных транслокаций на основе массового параллельного секвенирования обогащенных 3С-библиотек
Е. А. Можейко 1, В. С. Фишман 1, 2, *
1 Федеральный исследовательский центр Институт цитологии и генетики
Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
2 Новосибирский национальный исследовательский государственный университет
630099 Новосибирск, Россия
* E-mail: minja-f@ya.ru
Поступила в редакцию 07.05.2019
После доработки 06.06.2019
Принята к публикации 10.06.2019
Аннотация
Диагностика и лечение пациентов с наследственными заболеваниями требуют создания эффективных методов исследования индивидуальных геномов. Существующие подходы либо нацелены на поиск узкого набора геномных вариантов, либо слишком дороги для применения в рутинной практике. Мы исследовали возможность детекции точечных мутаций и межхромосомных транслокаций при помощи секвенирования обогащенных 3С-библиотек. Мы показали, что, с точки зрения детекции вариантов в экзонах, обогащенные 3С-библиотеки являются более информативными, чем полногеномные библиотеки, но уступают полноэкзомным данным. При этом транслокации существенно изменяют профиль пространственных контактов хроматина, что позволяет эффективно детектировать такие перестройки при анализе обогащенных 3С-библиотек.
Геномная диагностика является одной из активно развивающихся областей современной медицины. Геном каждого человека содержит 4–5 млн точечных мутаций, а также 1–2 тысячи крупных структурных перестроек, затрагивающих в совокупности около 20 млн пар оснований [1]. Хотя большинство вариаций встречается в некодирующих областях, часть из них является клинически значимой [1] и используется для диагностики и выбора подходов к лечению наследственных и онкологических заболеваний [2].
На сегодняшний день диагностическая эффективность различных генетических тестов лежит в диапазоне 30–70% [3, 4]. Причина столь невысокой диагностической эффективности связана в первую очередь с принципиальными ограничениями используемых методов. Например, цитологическое кариотипирование позволяет детектировать только крупные, от 1 до 10 млн пар оснований (Mb), хромосомные перестройки; сравнительная геномная гибридизация обладает более высокой разрешающей способностью – десятки тысяч пар оснований, но не чувствительна к сбалансированным перестройкам и не может детектировать однонуклеотидные замены [5]; секвенирование генных панелей и метод мультиплексной лигазозависимой амплификации (MLPA) позволяют детектировать точечные мутации, но только для ограниченного набора генов-кандидатов. Выбор одного из этих методов, относящихся к генетическим тестам “первой линии” и рутинно применяемых в практике, требует от врача сформулированной a priori гипотезы о типе генетической мутации и ее клинической значимости для пациента. Однако сформулировать такую гипотезу часто бывает сложно или даже невозможно, поскольку многие генетические патологии являются редкими и плохо описанными, варьируют в проявлениях от пациента к пациенту и могут быть вызваны, при сходном фенотипе, различными типами нарушений в многочисленных генах.
В последнее время в клинику активно внедряются методы, позволяющие детектировать хромосомные перестройки и однонуклеотидные замены в масштабе всего генома – полноэкзомное и полногеномное секвенирование. Преимуществом первого подхода является возможность получить детальную информацию о белок-кодирующих участках генома и за счет селективного обогащения экзомными последовательностями сократить стоимость секвенирования. При этом экзомное секвенирование не позволяет проводить эффективный поиск структурных перестроек, в особенности сбалансированных, если их границы не проходят внутри экзонов [6]. В отличие от полноэкзомного секвенирования, полногеномный анализ позволяет с точностью до нуклеотида восстанавливать структуру хромосомных перестроек, как было показано, в частности, нашей группой [7]. Полногеномное секвенирование позволяет также детектировать однонуклеотидные замены в кодирующих и некодирующих областях генома, делая этот метод золотым стандартом геномной диагностики.
Оборотной стороной точности и чувствительности полногеномного секвенирования является его дороговизна [8]. Стоимость глубокого полногеномного секвенирования, необходимого для идентификации пациент-специфичных геномных вариантов, и в особенности структурных перестроек, столь высока [8], что делает этот анализ недоступным в повседневной врачебной практике. Поэтому в медицинской генетике сохраняется запрос на создание новых методов, доступных для рутинной диагностики, способных одновременно детектировать однонуклеотидные замены и хромосомные перестройки.
Высокая требовательность к глубине секвенирования при полногеномном анализе обусловлена тем, что для детекции перестроек информативными являются только последовательности вблизи точек хромосомных разрывов, на расстоянии менее 1 тпн – а значит, чтобы найти такие точки, для всего генома необходимо обеспечить высокое покрытие прочтениями. Если бы участки вдали от точек разрыва как-либо изменялись при перестройке, возможно было бы просеквенировать только часть генома с высоким покрытием, и на основании полученных данных сделать вывод о наличии и локализации перестройки.
Оказалось, что такой подход можно применить при анализе трехмерной архитектуры хроматина в клетках [9]. Известно, что взаимное расположение участков линейной молекулы ДНК сильно влияет на частоту пространственных контактов между окружающими локусами: зависимость между трехмерным расстоянием в пространстве ядра и “нуклеотидным” расстоянием в геномных координатах описывается степенной функцией во всех изученных типах клеток [10–12]. Это означает, что хромосомная перестройка оказывает эффекты не только на частоту контактов районов, непосредственно расположенных в точках хромосомных разрывов, но и изменяет паттерн трехмерных контактов широкой области вокруг границы перестройки [9].
Основываясь на этой закономерности, недавно был предложен ряд методов детекции хромосомных перестроек на основе анализа изменений трехмерной организации ядра. Для этого создаются 3С-библиотеки [9–12], которые секвенируют с низким покрытием, что позволяет детектировать изменения пространственных контактов, связанные с произошедшей перестройкой [13–17]. Эти методы оказались наиболее чувствительны к детекции транслокаций, поскольку частоты межхромосомных контактов существенно ниже, чем внутрихромосомных [9–12], и, следовательно, в перестроенном геноме наблюдается резкое увеличение числа пространственных контактов между транслоцированными участками. Неглубокое секвенирование с низким покрытием каждого участка генома компенсируется тем, что информативной является протяженная область в несколько Mb, трехмерные контакты которой изменяются вследствие перестройки. При этом секвенирование с низким покрытием позволяет сделать методы детекции хромосомных перестроек, основанные на технологии 3С, относительно дешевыми (сравнимыми с ценой экзомного секвенирования). В то же время такой подход, в отличие от полноэкзомного или полногеномного секвенироваия, не дает возможности уверенно детектировать точечные мутации, что особенно актуально для последовательностей экзонов.
В данной работе мы предложили совместить гибридизационные методы целевого обогащения, используемые в протоколах экзомного секвенирования, с технологией 3C для одновременной детекции однонуклеотидных замен в целевых районах генома и плоногеномной детекции транслокаций. Используя данные, полученные на клетках K562 с известными хромосомными перестройками, мы показали, что секвенирование с глубиной ~100 млн парных прочтений позволяет уверенно идентифицировать транслокации. При этом, согласно сделанной нами оценке, секвенирование ~18 млн парных прочтений дает возможность достичь в среднем более чем 20-кратного (×20) покрытия экзомных последовательностей. Таким образом, метод целевого обогащения 3С-библиотек может быть эффективным инструментом для одновременного поиска однонуклеотидных замен в экзонах и межхромосомных транслокаций.
МАТЕРИАЛЫ И МЕТОДЫ
Для анализа данных 3С с обогащением последовательностями промоторов длинных некодирующих РНК из работы [18] были взяты данные в формате fastq для клеточных линий человека K562 и H1. С помощью программного обеспечения distiller (https://github.com/mirnylab/distiller-nf) мы провели выравнивание прочтений на геном человека версии hg19, отфильтровали ПЦР-дупликаты и отфильтровали неуникально картированные прочтения, которые характеризовались критерием MAPQ меньше 30.
Для визуализации 3C-матрицы контактов были построены с минимальным разрешением 5 тпн с помощью Juicer Tools (https://github.com/aidenlab/juicer/wiki/Pre) и сохранены в .hic формате. Далее .hic‑файлы были визуализированы с помощью Juicebox (https://github.com/aidenlab/Juicebox).
Кариотипы клеток H1 и K562 подробно описаны в статьях [19] и [20]. Стоит отметить, что клетки H1 имеют нормальный диплоидный кариотип, в то время как клетки K562 имеют гипотриплоидный кариотип и характеризуются множеством транслокаций. Координаты перестроек были конвертированы в геномные координаты hg19 с помощью инструмента USCS liftover.
Анализ проводился на базе вычислительного кластера ЦКП ИЦиГ СО РАН (Бюджетный проект № 0324-2019-0041) с использованием стандартных пакетов языка R.
РЕЗУЛЬТАТЫ
Дизайн эксперимента
Схема эксперимента, позволяющего совместить технологию 3С с экзомным обогащением, представлена на рис. 1. На первом этапе проводится приготовление стандартной 3С-библиотеки. Мы рекомендуем использовать для этого протокол in situ Hi-C, предложенный группой Liberman-Aiden в 2015 г. [11] и использованный нами [21] и другими группами [22] для анализа трехмерной организации генома. Технология in situ Hi-C заключается в фиксации хроматина формальдегидом, рестрикции ДНК в составе фиксированного хроматина и затем лигировании образовавшихся концов ДНК. Поскольку лигирование происходит в фиксированных ядрах (или их крупных фрагментах), диффузия ДНК существенно ограничена, и лигированными оказываются концы ДНК, которые были сближены в клетке. Поэтому количественный анализ продуктов лигирования при помощи секвенирования нового поколения позволяет получить информацию о трехмерной близости произвольной пары локусов генома.
Рис. 1.
Совмещение технологии 3С и экзомного обогащения для детекции транслокаций. Вверху схематично изображена 3D-конформация нормального (слева) и перестроенного (справа) генома. В качестве примера перестройки приведена реципрокная транслокация между хромосомами 1 (синий цвет) и 2 (красный цвет). Внизу в центре показаны полная и обогащенная экзомными последовательностями библиотеки, каждая полоска соответствует одной химерной молекуле ДНК, образовавшейся после лигирования. Справа и слева от них изображены тепловые карты пространственных контактов, соответствующие обогащенным библиотекам: каждой паре локусов соответствует одна точка на тепловой карте, а ее цвет отражает количество прочтений, содержащих фрагменты ДНК, картирующиеся на данную пару локусов. Красные полосы отражают районы обогащения (экзоны). Видно, что хромосомная перестройка приводит не только к изменению картины в месте соединения фрагментов двух хромосом, но и паттерна контактов, значительно удаленных от места перестройки.
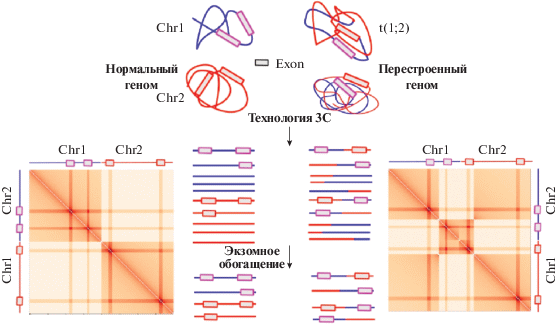
В оригинальном протоколе in situ Hi-C проводится секвенирование всей полученной 3С-библиотеки. Однако мы предлагаем перед секвенированием провести гибридизацию 3С-библиотеки с экзомными пробами. Подобные подходы уже применялись ранее для анализа трехмерной организации специфических локусов. В частности, в статье [18] 3С-бибилиотеки из клеток миелоидного лейкоза линии K562 и эмбриональных клеток линии H1 были обогащены последовательностями промоторов длинных некодирующих РНК за счет гибридизации с биотин-мечеными пробами, комплементарными целевым участкам. В работе [18], как и в других работах, использующих обогащение 3С-библиотек специфическими последовательностями, авторы не проводили анализ хромосомных перестроек – целью работы было исследование промотор-энхансерных взаимодействий для генов длинных некодирующих РНК. Между тем полученные данные представляют собой замечательную модель для оценки эффективности методов поиска перестроек на основе секвенирования 3С-библиотек, поскольку клетки K562 несут целый ряд транслокаций, недавно охарактеризованных в статье [23], в то время как клетки H1 имеют существенно более стабильный кариотип без цитологически выявляемых транслокаций [24] и поэтому могут служить контролем.
Примечательно, что в работе [18] для рестрикции ДНК в ходе приготовления 3С-библиотек использовался фермент ДНКаза I. Обычно для приготовления 3С-библиотек используются четырехбуквенные эндонуклеазы рестрикции [10–17]. Однако в этом случае около 50% всего генома будет расположено на расстоянии менее 400 пар нуклеотидов от какого-либо сайта рестрикции. Таким образом, при секвенировании библиотеки с размером вставки 300–400 пн (характерный размер вставки для библиотеки Illumina) возможно исследовать около 50% потенциальных сайтов однонуклеотидных замен в геноме. При использовании ДНКазы I, которая не имеет специфического сайта узнавания, можно анализировать фрагменты генома независимо от их удаленности от какого-либо сайта рестрикции и, таким образом, довести теоретическое количество анализируемых однонуклеотидных замен до 100%.
Таким образом, данные, полученные в работе [18], представляют удобную модель для детекции транслокаций на основе секвенирования обогащенных 3С-библиотек. Мы провели анализ этих данных, чтобы оценить эффективность поиска точечных мутаций и транслокаций в обогащенных 3С-библиотеках.
Анализ данных обогащенных 3С-библиотек клеток K562 и H1
Несмотря на то, что в ходе гибридизации со специфическими пробами происходит обогащение целевыми последовательностями, мы также детектировали значительное количество прочтений, картируемых на нецелевые области в данных, полученных для библиотек H1 и K562 (рис. 2,а). Можно привести две причины, объясняющие наличие таких прочтений. Во-первых, прочтения, относящиеся к нецелевым областям, возникают из-за контактов целевых областей с нецелевыми, при этом соответствующие этим прочтениям фрагменты ДНК, вероятно, гибридизовались с целевой пробой одним концом. Во-вторых, такие прочтения возникают при неспецифической гибридизации или неполной отмывке негибридизованных продуктов и в этом случае могут содержать контакты двух нецелевых областей.
Рис. 2.
Особенности данных обогащенных 3C-библиотек. а – участок первой хромосомы в клетках H1, представленный в виде тепловой карты матрицы контактов, визуализирован при помощи программного обеспечения JuiceBox. Для сравнения показаны внутрихромосомные контакты целевых областей с геномом (выше диагонали) и все детектированные контакты (ниже диагонали); б – распределение количества контактов произвольных геномных локусов размером 1 Mb в зависимости от расстояния между ними для клеток человека линии H1. Указано также распределение межхромосомных контактов 4-й и 13-й хромосомы H1.

Важной характеристикой 3С-данных является функция p(s), описывающая зависимость вероятности контакта между двумя локусами (p) от расстояния между этими локусами в линейной молекуле ДНК (s). Как и ожидалось, мы наблюдали быстрое падение частоты контактов при уменьшении геномного расстояния (рис. 2,б). Это позволяет предположить, что хромосомные перестройки будут приводить к резкому изменению частот контактов, связанному с изменениями геномного расстояния. При этом наличие контактов между нецелевыми областями важно для идентификации точек хромосомных перестроек, поскольку при прохождении перестройки вблизи нецелевого района, даже при небольшом покрытии этого района прочтениями, возможно будет детектировать сильные отклонения в частотах пространственных контактов.
Детекция межхромосомных транслокаций
При межхромосомных транслокациях область возле точки разрыва перестроенного региона одной хромосомы показывает высокие частоты контактов с областью около точки разрыва другой хромосомы. Эти частоты контактов по своей величине равны частотам внутрихромосомных контактов соседних локусов, которые сильно отличаются от частот межхромосомных контактов (рис. 2,б). Поэтому аномальные превышения частот контактов, возникающие при перестройке, хорошо заметны на тепловой карте контактов как для обогащенных 3С-библиотек, так и для 3С-библиотек, сделанных по протоколу in situ Hi-C (рис. 3,а, б).
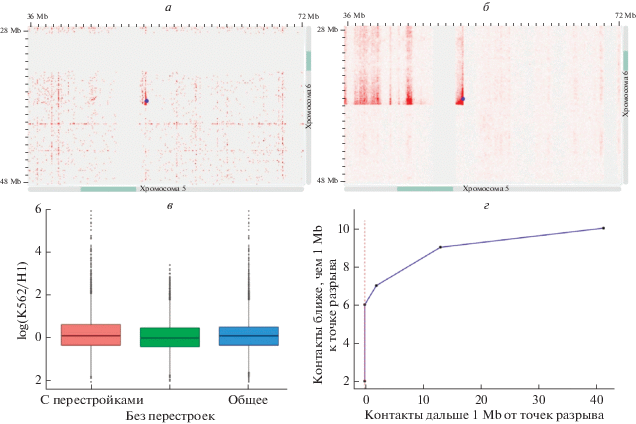
Рис. 3.
Идентификация перестроек по данным обогащенных 3C-библиотек. а, б – участок межхромосомных контактов хромосом 5 и 6 клеточной линии K562. На этом участке видны последствия транслокации. Данные из [18] (а) и данные из [10] (б). Синяя точка указывает на координаты контакта, сопоставленного с перестройкой, т.е. отношение K562/H1 выше заданного порога. в – диаграммы размаха логарифма распределения отношения частот контактов. Распределения логарифма отношений частот межхромосомных контактов хромосом с известной транслокацией (красный), хромосом без известных перестроек (зеленый) и их объединение (синий). Красная линия обозначает порог, после которого предполагается, что контакты принадлежат перестройкам. г – зависимость количества контактов в районах перестроек (т.е. не дальше 1 Mb от известных перестроек) и контактов не в районах перестроек от порога. Рассматривались межхромосомные контакты между хромосомами 3 и 10, 5 и 6, 12 и 21 и соответствующие им перестройки (t(5;6)(5pter → 5p?11::?), t(3;10)(10pter → 10q23::3p21 → 3pter), t(12;21)(21qte → rq11::12p11 → 12qter)). Порог увеличивается справа налево на одно стандартное отклонение общего распределения (синего, график в), начиная с пяти стандартных отклонений. Красная вертикальная линия показывает предполагаемый порог (восемь стандартных отклонений), позволяющий отличать отношения контактов, указывающих на перестройки.
Сложность систематического поиска аномально высоких межхромосомных контактов заключается в том, что отклонения частоты контактов от средней могут быть вызваны не только хромосомной перестройкой, но и особенностями конкретных геномных районов. Например, наличие повторов влияет на картируемость прочтений, а GC-состав – на эффективность амплификации фрагментов, что в конечном счете сказывается на числе контактов, детектируемых для данного района. Многие работы, посвященные анализу данных 3С и поиску хромосомных перестроек, предлагают проведение нормализации, учитывающей локус-специфические факторы, перед поиском аномально высоких контактов [16, 17, 23]. Наиболее распространенный способ нормализации – итеративная коррекция (Iterative Correction) [25]. Однако этот метод не подходит для нормализации данных секвенирования обогащенных 3С-библиотек, так как предполагает равномерное покрытие генома прочтениями, что не может выполняться после обогащения библиотеки целевыми последовательностями. На данный момент, насколько известно авторам, никем не предложен способ нормализации подобных данных.
Поскольку факторы отклонения зависят в первую очередь от особенностей конкретных геномных локусов (GC-состава, наличия повторов, эффективности амплификации и т.п.), можно допустить, что эти факторы для каждого локуса одинаковы у двух различных 3С-библиотек. В таком случае частота контактов одной и той же пары локусов, измеренная в двух 3С-экспериментах, будет одинаковой. Мы использовали это предположение при анализе контактов в 3С-экспреиментах для клеток H1 и K562.
Мы разделили весь геном на последовательно расположенные локусы размером 1 Mb, и для каждой пары локусов рассчитали частоту пространственных контактов, получив, таким образом, матрицу 3С-контактов для клеток K562 и H1. Пользуясь предположением о том, что факторы, влияющие на отклонения в числе контактов, не являются клеточно-специфичными, мы поделили частоты контактов 3C-матрицы K562 на частоты контактов матрицы H1. Распределение было умножено на константу, чтобы выборочное среднее для полученных отношений равнялось 1, тем самым была учтена разница в глубине секвенирования библиотек H1 и K562. Для удобства анализа был взят логарифм распределения. Таким образом, было получено распределение логарифмов отношений частот контактов для межхромосомных взаимодействий (рис. 3,в). Важно, что при таком анализе, принимая описанные выше допущения, отклонение значений логарифма отношения от нуля не может быть связано с особенностями конкретных локусов.
Поскольку для клеток К562 описан криотип, среди всех межхромосомных контактов, наблюдаемых в этих клетках, можно выделить те, в которых участвуют хромосомы с известными транслокацями. Так как вероятность контакта соседних локусов на одной хромосоме существенно больше вероятности контакта между хромосомами, то контакт локусов рядом с границей транслокации будет иметь гораздо более высокую частоту, чем контакт между той же парой локусов в клетках без транслокации. Это утверждение эквивалентно тому, что отношение частот контактов двух типов клеток будет отличаться от единицы, а логарифм – отличаться от нуля. Следует отметить, что клетки линии H1 не имеют описанных транслокаций, в то время как в клетках K562 описано 18 межхромосомных транслокаций [23]. Поэтому можно ожидать, что отношение значительного количества частот межхромосомных контактов K562 к H1 будет больше единицы. Это соответствует положительному значению логарифма, что и наблюдалось в полученных распределениях – медианное значение логарифма отношения частот контактов для хромосом, участвующих в транслокациях в клетках К562, оказалось выше, чем медианное значение логарифма для хромосом без перестроек (p-value 2.2 × 10–16). При этом медианное значение логарифма отношения частот контактов для хромосом без перестроек было равно нулю, т.е. в отсутствие перестроек средняя частота межхромосомных контактов в клетках H1 и K562 была одинаковой.
Различия в распределении частот межхромосомных контактов неперестроенных хромосом и хромосом с транслокациями в клетках К562 позволяют предположить, что существует способ систематически идентифицировать границы транслокаций. Очевидно, что такой подход должен заключаться в поиске контактов, частота которых существенно отличается от среднего значения частот межхромосомных взаимодействий, характерных для большого количества неперестроенных областей. При этом определенную сложность представляет выбор конкретного порогового значения отличия частоты контакта от средней, при превышении которого можно считать, что наблюдаемый контакт описывает границы транслокации. Сложность в выборе порога заключается в том, что частоты контактов локусов ДНК зависят не только от их линейного расстояния в геноме, но и от других биологических причин (например, фазовой сепарации эу- и гетерохроматина), а также технических факторов, часть из которых уже была перечислена выше (GC-состав локуса, локус-специфичная эффективность ферментативных реакций и т.п.). Часть из этих факторов была учтена нами в ходе нормализации – при переходе от абсолютных значений частот контактов в данном типе клеток к отношению частот контактов, наблюдаемых для двух типов клеток. Однако часть факторов – например, являющихся специфичными для определенного типа клеток или представляющих собой невоспроизводимые технические погрешности конкретного эксперимента (“шум”), – продолжают влиять на частоты контактов, что приводит к отклонениям отношения частоты межхромосомных контактов в двух типах клеток (K562 и H1) от единицы для большинства пар локусов, даже если они не находятся вблизи границ транслокаций. Таким образом, задача выбора такого порога отклонения отношения частоты контактов от единицы, при котором будут идентифицированы только контакты границ транслокаций, является нетривиальной.
Для решения этой задачи мы предприняли следующий эксперимент. Мы разделили все межхромосомные контакты генома человека на три группы. В первую группу (назовем ее “участки, достоверно содержащие границы транслокаций”) попали контакты локусов, расположенных не далее чем 1 Mb от границ транслокаций в клетках К562, при условии, что геномные координаты границ транслокаций были определены ранее в статье [23] с точностью не менее 1 Mb. Следует отметить, что в клетках К562 всего три такие транслокации (t(5;6)(5pter → 5p?11::?), t(3;10)(10pter → → 10q23::3p21 → 3pter), t(12;21)(21qte → → rq11::12p11 → 12qter)), представленные десятью контактами.
Во вторую группу (назовем ее “неопределенные участки”) попали контакты локусов, расположенных на расстоянии 1–5 Mb от границ известных транслокаций в клетках К562, для которых ранее была точно (точность не менее 1 Mb) определена граница, а также все межхромосомные контакты локусов, лежащих на хромосомах, для которых было показано наличие транслокаций, но границы транслокаций не были точно определены.
Наконец, в третью группу (назовем ее “участки, достоверно не содержащие транслокаций”) попали все остальные межхромосомные контакты. Геномные расстояния 1 и 5 Mb, использованные в данном анализе, продиктованы следующими соображениями. Во-первых, размер локусов, на которые был разбит геном для подсчета частот 3D-контактов, составлял 1 Mb, поэтому точность детекции границ транслокаций не могла быть выше этого значения. Во-вторых, согласно данным, представленным на рис. 2,б, частоты пространственных контактов быстро падают при увеличении геномного расстояния вплоть до 5 Mb, а контакты на большем геномном расстоянии крайне редки и, следовательно, малоинформативны для поиска перестроек. Нужно отметить, что изменение константы 5 Mb в большую или меньшую сторону не влияло на результаты анализа (данные не приведены).
Разбив все контакты на три вышеперечисленные категории, мы последовательно перебирали различные значения порога отличий частот контактов от среднего по геному, чтобы найти величину порога, при которой наиболее точно отделяются контакты границ транслокаций (принадлежат первой категории) от контактов участков, достоверно не содержащих границы транслокаций (третья категория). Для каждого значения порога мы считали, что все межхромосомные контакты, у которых отношение частот в клетках K562 (несущих транслокации) и H1 (не имеющих перестроек) выше значения порога, относятся к первой категории, а те контакты, для которых отношение ниже порога, – к третьей. Если часть контактов оказывалась отнесенной при таком вычислении к первой категории ошибочно, мы увеличивали значение порога на величину, равную одному стандартному отклонению выборки всех отношений частот межхромосомных контактов.
Результаты, представленные на рис. 3,г, показывают, что для порога, равного восьми стандартным отклонениям выборки всех отношений частот межхромосомных контактов, все контакты, не относящиеся к первой категории, оказываются ниже порога. При этом выше порога оказываются шесть из десяти контактов, относящихся к первой категории. Важно, что все три транслокации клеток К562, участвовавшие в анализе, оказываются представленными среди шести контактов, характеризующихся значениями выше порога. Таким образом, оказалось возможным выбрать пороговое значение отклонения частоты межхромосомных контактов от средней по геному, при котором выделяются исключительно контакты вблизи границ транслокаций.
ОБСУЖДЕНИЕ
Мы показали, что 3С-данные, обогащенные целевыми последовательностями, могут быть использованы для поиска различных геномных вариантов. Секвенирование с относительно небольшой глубиной (~18 млн парных прочтений) позволяет добиться минимального среднего покрытия (×20), необходимого для поиска точечных мутаций [26]. Безусловно, предпочтительным является более глубокое секвенирование (~100–150 млн парных прочтений), позволяющее достичь рекомендуемого для полноэкзомных и полногеномных технологий покрытия (более ×30–×50, [26]). Таким образом, с точки зрения поиска вариантов в экзонах представленный метод оказывается более дорогим, чем полноэкозомное секвенирование (при экзомном секвенировании среднее покрытие составляет ×50–×100 при глубине ~70 млн прочтений [27]), но более дешевым, чем полногеномное секвенирование (покрытие ×30 при секвенировании ~350 млн парных прочтений, [7]).
При этом полученные данные позволяют детектировать хромосомные транслокации. В этой работе мы сосредоточились на поиске трех гетерозиготных транслокаций в клетках К562, для которых была описана точная (с погрешностью <1Mb) граница. Все три были успешно найдены на разрешении 1 Mb. В будущем необходимо оценить, какое минимальное разрешение доступно для анализа подобных данных и, соответственно, с какой точностью могут быть с его помощью установлены границы транслокаций и какой минимальный размер участка, который можно зафиксировать данным методом. Еще одним важным вопросом является то, могут ли быть обнаружены с помощью подобных методов внутрихромосомные перестройки – инверсии, делеции и дупликации.
Наконец, нужно отметить, что у 3С-опосредованных методов есть широкий потенциал к дальнейшему развитию в качестве инструмента геномной диагностики. Во-первых, возможно использовать производные 3С-методов, например 4С [28], для поиска точечных мутаций и структурных перестроек в небольшой панели целевых генов. Во-вторых, помимо косвенных данных о перестройках, результаты секвенирования обогащенных 3С-библиотек несут прямую информацию о трехмерной организации генома. Эта информация представляет безусловный интерес для врачей-генетиков в контексте нарушений промотор-энхансерных взаимодействий, которые, как было убедительно показано в последние годы, могут являться причиной развития тяжелых патологий [9].
Работа выполнена при поддержке гранта РФФИ, грант № 18-29-13021.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Auton A., Brooks L.D., Durbin R.M. et al. A global reference for human genetic variation. // Nature. 2015. V. 526. № 7571. P. 68–74.
Clark M.M., Amber H., Sergey B. et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation // Sci. Transl. Med. 2019. V. 11. № 489. P. eaat6177. https://doi.org/10.1126/scitranslmed.aat6177
Shashi V., McConkie-Rosell A., Rosell B. et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders // Genet. Med. 2014. V. 16. № 2. P. 176–182. https://doi.org/10.1038/gim.2013.99
Levy B., Stosic M., Giordano J., Wapner R. Chromosomal microarrays and exome sequencing for diagnosis of fetal abnormalities // Hum. Reprod. Prenatal Genet. 2018. P. 577–595. https://doi.org/10.1016/b978-0-12-813570-9.00026-7
Miller D.T., Adam M.P., Aradhya S. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies // Am. J. Hum. Genet. 2010. V. 86. № 5. P. 749–764. https://doi.org/10.1016/j.ajhg.2010.04.006
D’Aurizio R., Pippucci T., Tattini L. et al. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2 // Nucl. Acid. Res. 2016. V. 44. № 20. P. e154. https://doi.org/10.1093/nar/gkw695
Gridina M.M., Matveeva N.M., Fishman V.S. et al. Allele-specific biased expression of the CNTN6 gene in IPS cell-derived neurons from a patient with intellectual disability and 3p26.3 microduplication involving the CNTN6 gene // Mol. Neurobiol. 2018. V. 55. № 8. P. 6533–6546. https://doi.org/10.1007/s12035-017-0851-5
Schwarze K., Buchanan J., Taylor J.C., Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature // Genet. in Med. 2018. V. 20. № 10. P. 1122–1130. https://doi.org/10.1038/gim.2017.247
Fishman V.S., Salnikov P.A., Battulin N.R. Interpreting chromosomal rearrangements in the context of 3-dimentional genome organization: A practical guide for medical genetics // Biochemistry (Mosc). 2018. V. 83. № 4. P. 393–401. https://doi.org/10.1134/S0006297918040107
Rao S.S., Huntley M.H., Durand N.C. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping // Cell. 2014. V. 159. № 7. P. 1665–1680. https://doi.org/10.1016/j.cell.2014.11.021
Lieberman-Aiden E., van Berkum N.L., Williams L. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome // Science. 2009. V. 326. № 5950. P. 289–293. https://doi.org/10.1126/science.1181369
Battulin N., Fishman V.S., Mazur A.M. et al. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach // Genome Biol. 2015. V. 16. P. 77. https://doi.org/10.1186/s13059-015-0642-0
Korbel J.O., Lee C. Genome assembly and haplotyping with Hi-C // Nat. Biotechnol. 2013. V. 31. № 12. P. 1099–1101. https://doi.org/10.1038/nbt.2764
Burton J.N., Adey A., Patwardhan R.P. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions // Nat. Biotechnol. 2013. V. 31. P. 1119–1125. https://doi.org/10.1038/nbt.2727
Kaplan A., Ledford D., Ashby M. et al. Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy // J. Allergy Clin. Immunol. 2013. V. 132. № 1. P. 101–109. https://doi.org/10.1016/j.jaci.2013.05.013
Harewood L., Kishore K., Eldridge M.D. et al. Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours // Genome Biol. 2017. V. 18. P. 125. https://doi.org/10.1186/s13059-017-1253-8
Chakraborty A., Ay F. Identification of copy number variations and translocations in cancer cells from Hi-C data // Bioinformatics. 2017. https://doi.org/10.1093/bioinformatics/btx664
Ma W., Ay F., Lee C. et al. Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes // Nat. Methods. 2014. V. 12. № 1. P. 71–78. https://doi.org/10.1038/nmeth.3205
Chen Y., Dejosez M., Zwaka T.P., Behringer R.R. H1 and H9 human embryonic stem cell lines are heterozygous for the ABO locus // Stem Cells Dev. 2008. V. 17. № 5. P. 853–855. https://doi.org/10.1089/scd.2007.0226
Naumanna S., Reutzela D., Speicherb M., Decker H. Complete karyotype characterization of the K562 cell line by combined application of G-banding, multiplex-fluorescence in situ hybridization, fluorescence in situ hybridization, and comparative genomic hybridization // Leuk. Res. 2001. V. 25. № 4. P. 313–322. https://doi.org/10.1016/S0145-2126(00)00125-9
Fishman V., Battulin N., Nuriddinov M. et al. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes' chromatin // Nucl. Acid. Res. 2018. V. 47. № 2. P. 648–665. https://doi.org/10.1126/scitranslmed.aat6177
Ma W., Ay F., Lee C. et al. Using DNase Hi-C techniques to map global and local three-dimensional genome architecture at high resolution // Methods. 2018. V. 142. P. 59–73. https://doi.org/10.1016/j.ymeth.2018.01.014
Dixon J.R., Xu J., Dileep V. et al. Integrative detection and analysis of structural variation in cancer genomes // Nat. Genet. 2018. V. 50. № 10. P. 1388–1398. https://doi.org/10.1038/s41588-018-0195-8
Tosca L., Feraud O., Magniez A. et al. Genomic instability of human embryonic stem cell lines using different passaging culture methods // Mol. Cytogenet. 2015. V. 8. P. 30. https://doi.org/10.1186/s13039-015-0133-8
Imakaev M., Fudenberg G., McCord R.P. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization // Nat. Methods. 2012. V. 9. № 10. P. 999–1003. https://doi.org/10.1038/nmeth.2148
Sims D., Sudbery I., Ilott N.E. et al. Sequencing depth and coverage: key considerations in genomic analyses // Nature Rev. Genetics. 2014. V. 15. P. 121–132. https://doi.org/10.1038/nrg3642
Shigemizu D., Momozawa Y., Abe T. et al. Performance comparison of four commercial human whole-exome capture platforms // Sci. Reports. 2015. V. 5. № 12742. https://doi.org/10.1038/srep12742
Splinter E., de Wit E., van de Werken H.J. et al. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation // Methods. 2012. V. 58. № 3. P. 221–230. https://doi.org/10.1016/j.ymeth.2012.04.009
Дополнительные материалы отсутствуют.