

Генетика, 2019, T. 55, № 5, стр. 583-592
Молекулярно-генетические причины и клиническое описание бранхио-ото-ренального синдрома
О. Л. Миронович 1, *, Е. А. Близнец 1, Т. Г. Маркова 2, Н. Н. Алексеева 2, Т. И. Голубева 3, О. П. Рыжкова 1, А. В. Поляков 1
1 Медико-генетический научный центр
115478 Москва, Россия
2 Российский научно-практический центр аудиологии и слухопротезирования
Федерального медико-биологического агентства России
117513 Москва, Россия
3 Северодвинская городская детская клиническая больница
164504 Северодвинск, Россия
* E-mail: mironovich_333@mail.ru
Поступила в редакцию 18.06.2018
После доработки 04.09.2018
Принята к публикации 24.09.2018
Аннотация
Бранхио-ото-ренальный (БОР) синдром – аутосомно-доминантное заболевание, характеризующееся сочетанием нарушения слуха с преаурикулярными ямками, шейными свищами или кистами и аномалиями почек различного типа. Мутации в гене EYA1 обуславливают до 40% случаев БОР-синдрома. В данной работе впервые представлено подробное клиническое описание и молекулярно-генетическое исследование БОР-синдрома в Российской Федерации среди восьми российских пациентов из четырех неродственных семей. У трех пациентов обнаружены ранее не описанные патогенные варианты (c.394C>T (p.Gln132*), c.519delT (p.Gln174Asnfs*66), c.1360G>A (p.Gly454Ser)) в гене EYA1. Так же проведен анализ впервые выявленного в более раннем исследовании патогенного варианта c.858delC (p.Ile286Leufs*79). Результаты, полученные в настоящей статье, демонстрируют значительный вклад генетической патологии, обусловленной мутациями в гене EYA1, в заболеваемость БОР-синдромом у российских больных. Частота встречаемости клинических признаков больших диагностических критериев БОР-синдрома у российских пациентов соответствует зарубежным данным.
Бранхио-ото-ренальный синдром (БОР-синдром, синдром Мельника–Фрейзера) – редкое наследственное аутосомно-доминантное заболевание, характеризующееся проявлениями дизэмбриогенеза производных жаберных дуг (околоушные ямки и шейные свищи или кисты, пороки развития наружного и среднего уха) зачастую в сочетании с патологией почек и/или иных органов [1, 2]. Впервые подробно признаки синдрома описали американский генетик M. Melnick в 1975 г. [3] и канадский генетик F.C. Fraser в 1978 г. [2]. Сегодня для постановки клинического диагноза используются большие и малые диагностические критерии, предложенные Chang с соавт. в 2004 г. [4]. Большие критерии включают наиболее частые при синдроме аномалии: потерю слуха, преаурикулярные ямки, аномалии почек (гипоплазия/агенезия, поликистоз, ротация почек, аномалии чашечно-лоханочной системы), свищи шеи и стеноз наружного слухового канала. Малые критерии включают аплазию слезного протока, расщелину неба, ретрогнатию, врожденную дисплазию тазобедренного сустава, паралич лицевого нерва, слезотечение (gustatory lacrimation) и кисты поджелудочной железы. Наличие трех больших критериев или сочетание двух больших и двух малых критериев или двух больших критериев у пробанда и одного большого критерия у его родственника первой степени позволяет поставить клинический диагноз бранхио-ото-ренального синдрома. Точная распространенность БОР-синдрома неизвестна. В 1976 г. F.C. Fraser обследовал 3640 детей с глубоким нарушением слуха и обнаружил пять детей (0.15%) с семейной историей жаберных фистул и преаурикулярных ямок (1 : 700 000) [5]. Четыре года спустя F.C. Fraser обследовал 421 ребенка в монреальской школе для глухих, из них 2% имели клинические признаки БОР-синдрома [6]. Используя эти данные, F.C. Fraser оценил распространенность синдрома в 1 : 40 000. Истинная распространенность, вероятно, находится между этими двумя значениями. БОР-синдром клинически гетерогенен, имеет высокую пенетрантность и варьирующую экспрессивность всех признаков синдрома, даже у больных членов одной семьи [7].
БОР-синдром – генетически гетерогенное заболевание, известны три генетические формы синдрома. До 40% случаев БОР-синдрома представляют собой генетический тип 1 (OMIM#113650), который обусловлен мутациями в гене EYA1. Выделен в отдельную нозологическую единицу аллельный и фенотипический вариант БОР-синдрома типа 1 – бранхио-отический синдром (БО-синдром, OMIM#602588), характеризующийся всеми клиническими признаками БОР-синдрома, но без патологии почек [8]. Доминантные мутации в гене EYA1 приводят также к сходному заболеванию производных жаберных дуг – ото-фацио-цервикальному синдрому генетического типа 1 (OMIM#166780). У 2% больных БОР/БО-синдромом патогенные варианты обнаруживаются в гене SIX5 [9] (генетический тип 2, OMIM#610896), у 2.5% больных – в гене SIX1 (тип 3, OMIM#608389) [10].
Ген EYA1 представляет собой человеческий гомолог гена дрозофилы, нокаут которого приводит к полному отсутствию или пороку развития глаз (от англ. “eyes absent” – отсутствие глаз) [11]. В 1989 г. Haan с соавторами описали семью с клиническими признаками БОР-синдрома, у которых была выявлена транслокация на хромосоме 8q [12]. Благодаря данной находке местоположение гена было уточнено S. Abdelhak с соавторами посредством позиционного клонирования, в данной работе также выявлены первые мутации в гене [11]. Ген EYA1 состоит из 16 экзонов и охватывает область более чем в 150000 пн. Инициирующий транскрипцию ATG-кодон находитcя в экзоне 3. На сегодняшний день в мире описано около 200 мутаций в гене EYA1. Мутации располагаются на всем протяжении гена, “горячих” точек и экзонов в гене нет [13]. Стоит отметить, что взаимосвязи между типом мутаций и наличием или тяжестью клинических признаков не наблюдается. Y. Zhang c соавторами показали, что LOF-варианты (от англ. “loss of function” – потеря функции) в гене EYA1 не связаны с более тяжелым фенотипом по сравнению с миссенс-вариантами [14].
Ген SIX1 локализуется на хромосоме 14q23.1, охватывает область в 1376 пн и состоит из двух кодирущих экзонов; на данный момент в мире описано 16 мутаций в гене, при этом одна мутация c.328C>T (p.R110Q) выявлена в шести неродственых семьях в разных этнических группах [10]. Ген SIX5 локализуется на хромосоме 19q13.32, охватывает область в 3145 пн и состоит из трех кодирующих экзонов. На сегодняшний день зарегистрировано девять патогенных вариантов в данном гене.
Продуктом гена EYA1 является двухфункциональный белок (The Eyes Absent proteins) – член семейства белков EYA (EYA1–4), компонент консервативной регуляторной сети (Pax-Six-Eya-Dach (PSEDN)), играющей ключевую роль в развитии многих органов и систем, в частности в морфогенезе глаз, внутреннего уха, почек и жаберных дуг [15, 16]. Гены EYA экспрессируются в различных тканях на ранних этапах эмбриогенеза, и хотя у каждого гена своя уникальная картина экспрессии, существует ее обширное перекрытие. Например, исследования на мышах показали, что экспрессия EYA1, EYA2 и EYA4 выражена в пресомитной мезодерме и мезенхиме головы зародыша, но только EYA1 и EYA4 экспрессируются в отических пузырьках [17]. В развивающейся почке мыши картина экспрессии указывает на роль EYA1 в развитии метанефрических клеток [11]. Уникальной особенностью белков семейства EYA является то, что данные белки выполняют сразу несколько биохимических функций [18]. C-терминальный домен обладает тирозин-фосфатазной активностью и таким образом играет центральную роль в репарации ДНК [19, 20]. N-терминальный домен обладает опосредованной транскрипционной активностью: он не имеет собственной ДНК-связывающей функции, однако является коактиватором белковых продуктов генов семейства Six (SIX1, SIX2, SIX3, SIX4, SIX5, SIX6) – транскрипционных факторов, контролирующих экспрессию генов-мишеней [14]. Данные функции белка EYA1 объясняют его роль в регуляции транскрипции и передаче клеточных сигналов во время органогенеза.
Ранее в 2004 и 2006 гг. соавтором настоящей работы Т.Г. Марковой были опубликованы первые сообщения о российских пациентах с БОР-синдромом, в которых приведено клиническое описание трех неродственных пациентов, у одного из которых клинический диагноз был подтвержден на молекулярно-генетическом уровне – выявлена гетерозиготная мутация в гене EYA1 – c.858delC (NM_000503), или c.759delC (NM_172060) [1, 21]. На сегодняшний день в русскоязычной научной литературе других сообщений о данном синдроме нет. В настоящей работе приведено описание результатов клинического и молекулярно-генетического исследования еще четырех российских случаев БОР-синдрома, а также биоинформатического анализа клинического значения трех выявленных неописанных ранее мутаций в гене EYA1 и одной мутации, выявленной в более раннем исследовании [1, 21].
МАТЕРИАЛЫ И МЕТОДЫ
Пациенты. Проводилось клиническое обследование восьми пациентов с БОР-синдромом из четырех неродственных семей. Материалом для молекулярно-генетического исследования явились образцы ДНК четырех пробандов из данных семей, трех родственников с БОР-синдромом и двух здоровых родителей. Также в описание результатов работы для сравнения и обзора случаев включены данные исследования пациентов из ранней публикации Марковой [1].
Клиническое обследование. Все пациенты были клинически обследованы в Российском научно-практическом центре аудиологии и слухопротезирования (Москва). Характер и степень нарушения слуха оценивались с помощью тональной пороговой аудиометрии и тимпанометрии. Обследование так же включало осмотр врача генетика, сурдолога-отоларинголога, компьютерную томографию височной кости (для подтверждения наличия аномалий развития среднего и внутреннего уха) и ультразвуковое исследование почек и осмотр терапевта/нефролога.
Молекулярно-генетическое исследование. Выделение геномной ДНК из лейкоцитов периферической крови выполняли с помощью готового набора реактивов Wizard® Genomic DNA Purification Kit (Promega, США).
Образцы ДНК четырех пробандов исследованы с помощью таргетной MPS-панели “Hearloss” (от англ. hearing loss – потеря слуха), разработанной в лаборатории ДНК-диагностики МГНЦ. В связи с тем, что физические возможности панели были ограничены, в нее были включены 35 частых генов, ответственных за развитие несиндромальной потери слуха и синдромальных форм тугоухости (STRC, MYO7A, MYO15A, TECTA, SLC26A4, CDH23, USH2A, TMPRSS3, TMC1, COL11A2, OTOF, EYA1, OTOA, PCDH15, PAX3, KCNQ4, LOXHD1, WFS1, ADGRV1, MYH14, MYO6, ACTG1, PTPRQ, MYH9, OTOGL, TRIOBP, CLDN14, LRTOMT, DFNB59, TPRN, WHRN, ALMS1, POU3F4, SMPX, CHD7). Для пробоподготовки использовалась технология AmpliSeq™, представляющая собой ультрамультиплексную ПЦР [22]. Секвенирование проводилось на платформе Ion S5 (Life Technologies, США). Разработанная панель “Hearloss” протяженностью 329.89 тнп включает 1800 ампликонов размером от 125 до 275 пн. Расчетное покрытие кодирующих областей генов по данным разработчика (Ion AmpliSeq Designer) составляет 97.73%.
Обработка данных секвенирования проведена с использованием стандартного автоматизированного алгоритма, предлагаемого TermoFisher Scientific (Torrent Suite™), а также программного обеспечения Gene-Talk. Фильтрация выявленных вариантов нуклеотидной последовательности проводилась по популяционной частоте: не анализировались варианты с частотой более 1% согласно данным баз проектов 1000 Genomes Project, ESP6500 и Genome Aggregation Database. Интерпретация клинического значения выявленных вариантов нуклеотидной последовательности проводилась согласно “Руководству по интерпретации данных, полученных методами массового параллельного секвенирования (MPS)” [23]. Обозначение вариантов гена EYA1 осуществляли в соответствии с международной номенклатурой HGVS (http:// www.hgvs.org/mutnomen/), референсная последовательность мРНК: NM_000503.5. Частота описанных вариантов среди пациентов с БОР-синдромом и здоровых людей оценивалась с помощью баз данных HGMD® Professional 2017.4 (https://portal.biobase-international.com), 1000 Genomes Project (http://browser.1000genomes.org, http://www.ensembl.org), gnomAD (http://gnomad.broadinstitute.org) и NCBI ClinVar, dbSNP. Патогенность выявленных вариантов анализировали с применением интернет-ресурсов, предсказывающих эффекты мутаций: Mutation Taster (http://www.mutationtaster.org), PROVEAN, SIFT (http://provean.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), NetGene2 (http:// www.cbs.dtu.dk/services/NetGene2/) и SpliceFinder (http://www.umd.be/HSF3/).
Все кодирующие экзоны и прилегающие экзон-интронные соединения гена EYA1 полностью покрыты в панели (диапазон покрытия составлял от 10× до 700×). Некодирующий экзон 1 оказался полностью не покрыт. Образец ДНК пробанда, в котором не найдено патогенных и вероятно-патогенных вариантов, был исследован на наличие мутаций в экзоне 1 гена EYA1 методом прямого автоматического секвенирования по Сенгеру на приборе ABI Prism 3100 (Applied Biosystems). Наличие патогенных и вероятно-патогенных вариантов, выявленных у пробандов методом MPS, также было проверено данным методом. В качестве матрицы для секвенирования использовали фрагменты ДНК, полученные после проведения ПЦР с использованием оригинальных олигонуклеотидных праймеров (табл. 1), которые синтезировались в ЗАО “Евроген”. ПЦР проводили по стандартному протоколу, описанному ранее [24]. Температура отжига праймеров составила 62°С.
Таблица 1.
Последовательность праймеров для ПЦР
| Фрагмент EYA1 | Последовательность олигонуклеотидов, 5' → 3' |
|---|---|
| Экзон 1 | F: CCATGGGCAGACCTACAAGGC |
| R: CAGGACAGCTGTTGTCAGTCAC | |
| Экзон 5–6 | F: CCAAATATCTGAAATTTCATATGGCC |
| R: CACGCATGCCCATGCGATAAC | |
| Экзон 7 | F: CCAGCTTTTGAAAATGGACAGATAG |
| R: GCCTCTAAGCCCAATCCAGTTG | |
| Экзон 14 | F: GTTGAAAAATCCTCCAATTAAGGTGC |
| R: GGAATTGTATCTGGACTGTCTTAAC |
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Клиническое описание пациентов с БОР-синдромом
Клинически обследованы семь пациентов из трех семей и один спорадический случай с признаками БОР-синдрома. Большие клинические признаки синдрома, наблюдаемые у пациентов, представлены в табл. 2. В таблице также представлены дополнительно данные трех российских неродственных пациентов с признаками БОР-синдрома, опубликованные ранее Т.Г. Марковой [1]. Пример аудиограммы пробанда семьи 3 с БОР-синдромом представлен на рис. 1.
Таблица 2.
Клиническая характеристика пациентов с БОР-синдромом
| Клинические признаки | Семья 1 | Семья 2 | Семья 3 | Семья 4 | Семья 5** | Семья 6** | Семья 7** | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| пробанд* | дочь | сын | мать | пробанд* | пробанд* | пробанд* | сын | пробанд* | пробанд* | пробанд* | |
| Возраст на момент осмотра, лет | 38 | 8 | 6 | 30 | 8 | 8 | 25 | 3 | 30 | 20 | нд |
| Оттопыренные ушные раковины | – | + | + | + | + | + | + | + | + | – | – |
| Преаурикулярные ямки и кисты | |||||||||||
| двусторонние | + | + | + | + | + | + | + | + | + | + | |
| односторонние | + | ||||||||||
| Шейные кисты и свищи | |||||||||||
| двусторонние | + | + | + | – | |||||||
| односторонние | + | + | + | + | + | + | + | – | |||
| Кондуктивная (К)/смешанная (С)/нейросенсорная (Н) тугоухость двусторонняя | +C | +К | +К | +С | +С | +К | +С | +С | +H | +C | +H |
| Костно-воздушный разрыв | + | + | + | + | + | + | + | нд | нд | нд | нд |
| Степень тугоухости (левое/правое ухо) | III–IV | III | III | III–IV | III/I | I | II | I | III–IV | нд | нд |
| Аномалии, выявленные при КТ височных костей | +а | нд | +а | нд | +б | +в | нд | нд | нд | нд | нд |
| Патология почек | +г | – | – | +г | – | +д | +г | – | + д | – | – |
| Семейный анамнез | + | + | – | + | + | + | + | ||||
* Пробанд, для которого проведен поиск мутаций в гене EYA1. ** Данные трех российских неродственных пациентов с признаками БОР-синдрома, опубликованные ранее Т.Г. Марковой [1]. Примечание. “+” – признак присутствует; “–” – признак отсутствует; нд – нет данных, так как исследование не проводилось; “а” – широкий водопровод преддверия, фронтальный полукружный канал четко не визуализируется, внутренний слуховой проход расширен; “б” – барабанная полость неправильной формы, длинный отросток наковальни неправильной формы, истончен, улитка уменьшена в размерах, отмечается дисплазия горизонтального полукружного канала, расширен водопровод преддверия и ямки эндолимфатического мешка; “в” – аномалии развития слуховых косточек, длинный отросток наковальни неправильной формы, истончен, отмечается гипоплазия улитки и заднего полукружного канала, внутренний слуховой проход расширен и имеет диаметр 6 мм с обеих сторон; “г” – частые обострения пиелонефрита; “д” – агенезия правой почки.
Рис. 1.
Аудиограмма пробанда семьи 3 с бранхио-ото-ренальным синдромом. С обеих сторон зарегистрирован костно-воздушный интервал >20 дБ на низких частотах и на 8 кГц, свидетельствующий о наличии тугоухости кондуктивного типа.
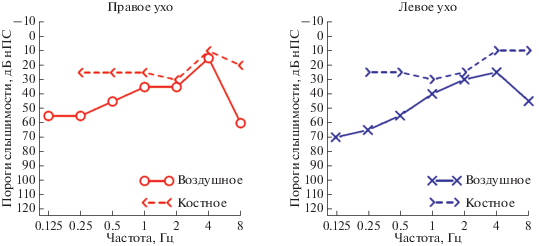
Согласно литературным данным нарушение слуха встречается у 93% пациентов с признаками БОР-синдрома. Данные представлены для молекулярно не подтвержденных выборок. В большинстве случаев наблюдается смешанный тип тугоухости (50%), реже – кондуктивный (30%) и сенсоневральный тип (20%) [25]. Возраст начала нарушений и степень потери слуха варьируют – от раннего детского до середины второго десятилетия жизни, от легкой до тяжелой и практически полной глухоты [1]. У большинства пациентов с БОР-синдромом тугоухость сочетается с аномалиями развития структур внутреннего уха – кохлеарной гипоплазией (63%), широким водопроводом преддверия (46%), луковицеобразным внутренним слуховым проходом (25%) и аномалиями косточек (50%) [25–27]. Преаурикулярные ямки наблюдаются в 70–80% случаев, чашеобразные уши отмечаются у 36% пациентов, шейные фистулы – у 63% пациентов [25, 27]. Данные о встречаемости аномалий почек у пациентов с БОР-синдромом варьируют от 15 до 65% по данным разных авторов [1, 28]. Встречаются как грубые пороки развития почек – такие как двусторонняя агенезия, приводящая к мертворождению, так и менее тяжелые аномалии – гипоплазия почек, гидронефроз, пузырно-мочеточниковый рефлюкс.
У шести исследуемых нами семей был диагностирован классический БОР-синдром с присутствием всех клинических признаков больших диагностических критериев, предложенных Chang с соавт. [4]. В семье 7 (см. табл. 2) наблюдалось сочетание преаурикулярных ямок и глухоты без других признаков синдрома. Таким образом, тугоухость наблюдалась у всех пациентов и степень нарушения слуха варьировала от легкой до тяжелой. Преаурикулярные ямки и свищи присутствовали у 10 пациентов (91%). В большинстве случаев наблюдался смешанный тип тугоухости (55%), реже – кондуктивный (27%) и сенсоневральный тип (18%). Не у всех пациентов было проведено КТ височных костей, однако у всех исследованных лиц выявлены изменения структур среднего и внутреннего уха, характерные для БОР-синдрома, такие как гипоплазия улитки и широкий водопровод преддверия. Патология почек наблюдалась реже других признаков: в 2 случаях (18%) – агенезия правой почти, в 3 случаях (27%) – частые обострения пиелонефрита, которые являются результатом пузырно-мочеточникового рефлюкса. Таким образом, клинические признаки и частота их встречаемости у российских пациентов с БОР-синдромом соответствует зарубежным данным.
Молекулярно-генетическое исследование пациентов с БОР-синдромом
Образцы ДНК четырех пробандов исследованы с использованием таргетной MPS-панели “Hearloss” (см. Материалы и методы). В результате у трех пациентов в гене EYA1 обнаружены гетерозиготные мутации: c.394C>T, c.519delT и c.1360G>A. Данные варианты нуклеотидной последовательности не были описаны ранее в литературе и не представлены в базах данных HGMD, dbSNP, 1000 Genomes Project и gnomAD (см. Материалы и методы). В табл. 3 показаны результаты проведенного биоинформатического анализа клинического значения как трех вариантов, выявленных в настоящей работе, так и варианта c.858delC, выявленного ранее у российского пациента в работе V. Migliosi в соавторстве с Т.Г. Марковой [1, 21].
Таблица 3.
Варианты гена EYA1, являющиеся причиной БОР-синдрома у российских пациентов
| Номер семьи пробанда | Название варианта | Локализация в гене | Эффект | Клиническое значение** | Критерии патогенности |
|---|---|---|---|---|---|
| 1 | c.519delT | Экзон 7 | p.Gln174Asnfs*66 | Патогенный | PVS1 + PM2 + PP4 |
| 2 | c.394C>T | Экзон 6 | p.Gln132* | » | PVS1 + PM2 + PP4 |
| 3 | c.1360G>A | Экзон 14 | p.Gly454Ser и/или нарушение сплайсинга | » | PS2 + PM1 + PM2 + + PP3 + PP4 |
| 5* | c.858delC | Экзон 10 | p.Ile286Leufs*79 | » | PVS1+ PM2 + PP4 |
Среди вариантов в гене EYA1, описанных ранее в литературе, 45 миссенс-мутаций, 36 точковых делеций, 22 точковые вставки, 5 точковых инделов, 29 нонсенс-мутаций, 3 мутации, приводящие к потере стоп-кодона, 30 мутаций сайтов сплайсинга, 25 протяженных делеций, 2 протяженные вставки и 6 сложных перестроек включены в базу данных HGMD ® Professional 2017.4 как “мутации, обуславливающие заболевание”. Таким образом, в базе HGMD зарегистрировано 116 нуль-вариантов (мутаций, обуславливающих образование преждевременного стоп-кодона, сдвиг рамки считывания, изменение канонических +/–1 или 2 сайтов сплайсинга) и 45 миссенс-мутаций. При этом по состоянию на январь 2017 г. в базе ClinVar в гене EYA1 среди доброкачественных (или вероятно-доброкачественных) вариантов не описано ни одного нуль-варианта и зарегистрировано 8 миссенс-замен. Эти данные демонстрируют, что нуль-варианты и миссенс-мутации в гене EYA1 являются распространенной причиной БОР/БО-синдрома, и частота доброкачественных мутаций указанных функциональных типов в гене EYA1 сравнительно низка. Поэтому при классификации обнаруженных неописанных нуль- и миссенс-вариантов применимы очень сильный PVS1 и поддерживающий PP2 критерии патогенности согласно рекомендациям для классификации патогенных вариантов [23].
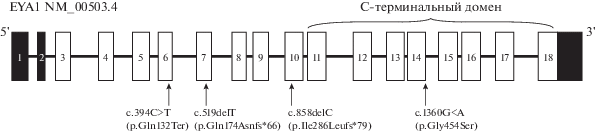
Три из анализируемых в настоящей работе варианта нуклеотидной последовательности – нонсенс-мутация c.394C>T (p.Gln132*) и приводящие к сдвигу трансляционной рамки считывания и образованию преждевременного стоп-кодона делеции c.519delT (p.Gln174Asnfs*66) и c.858delC (p.Ile286Leufs*79) – обусловливают отсутствие синтеза полноразмерного белка, т.е. являются нуль-мутациями. Четвертый вариант нуклеотидной последовательности, c.1360G>A (p.Gly454Ser), является миссенс-вариантом, который также может влиять на сплайсинг, поскольку затрагивает последний нуклеотид экзона 14 – донорный сайт сплайсинга. Все варианты локализуются, вероятно, в функционально значимых участках последовательности: анализ выровненных нуклеотидных и белковых последовательностей, гомологичных гену EYA1 и белку EYA1, показал эволюционную консервативность областей локализации мутаций среди почти всех классов животных (от червей до человека) (рис. 2). Белок EYA1 состоит из С-терминального домена, состоящего из 271 аминокислоты, охватывающего последние 8 экзонов белка (EYA-domain – ED), и N-терминального домена (N-terminal domain – NTD). C-терминальный домен является высококонсервативным – он гомологичен гену дрозофилы и его структура идентична в пределах семейства белков EYA [1, 19, 29]. N-терминальный домен (или eyaVR – от англ. “eya variable region”) является более вариабельным внутри семейства [21, 29]. Вариант c.1360G>A локализуется в экзоне 14 гена EYA1 и, таким образом, затрагивает С-терминальный домен, что указывает в пользу его патогенности. Три других мутации, приводящие к образованию преждевременного стоп-кодона, локализуются в экзонах 6, 7, 10 и вероятно приводят к образованию усеченного белка EYA1, у которого отсутствует ED-домен (рис. 3).
Рис. 2.
Анализ консервативности выявленных вариантов нуклеотидной последовательности в гене EYA1 с помощью программы MutationTaster.

Анализ эффекта in silico четырех анализируемых мутаций с помощью биоинформатического интернет-ресурса MutationTaster и миссенс-мутации дополнительно с помощью ресурсов PolyPhen2, PROVEAN, SIFT и UMD-predictor показал патогенный эффект всех мутаций с высокой достоверностью предсказания. Исключением является результат анализа миссенс-мутации с использованием ресурса PolyPhen-2, который показал нейтральный эффект с чувствительностью 0.93 и специфичностью 0.86. Программы, предсказывающие влияние на сплайсинг (NetGene2 и SpliceFinder), при анализе миссенс-мутации показали наличие нарушения процесса сплайсинга: SpliceFinder – исчезновение донорного сайта сплайсинга, NetGene2 – снижение вероятности использования истинного донорного сайта с 80% (при анализе нормальной последовательности) до 0% (при анализе последовательности с мутацией), т.е. исчезновение сайта. Отсутствие сплайсинга интрона 14, вероятно, приводит к вставке последовательности данного интрона длиной 962 пн в последовательность мРНК и сдвигу трансляционной рамки считывания с образованием преждевременного стоп-кодона. Для остальных трех мутаций программы NetGene2 и SpliceFinder показали незначительное влияние на сплайсинг или его отсутствие. Таким образом, биоинформатический анализ показал с высокой достоверностью, что все четыре исследуемых варианта гена EYA1 представляют собой нуль-мутации, приводящие к нарушению синтеза белка.
Наличие трех вариантов нуклеотидной последовательности гена EYA1, выявленных методом MPS, было подтверждено методом прямого автоматического секвенирования по Сэнгеру (рис. 4). Также на наличие данных вариантов нуклеотидной последовательности исследованы образцы ДНК членов семей пробандов. В семье 2 вариант c.394C>T (p.Gln132*) выявлен у пробанда, а также у сына и дочери пробанда с БОР-синдромом. В семье 1 вариант c.519delT (p.Gln174Asnfs*66) выявлен у пробанда и его матери с БОР-синдромом. В семье со спорадическим случаем заболевания (№ 3) вариант c.1360G>A (p.Gly454Ser) выявлен у пробанда и отсутствовал у его здоровых родителей. Таким образом, к данному варианту стало возможным применение сильного критерия патогенности PS2 (de novo вариант).
Рис. 4.
Родословные, генотипы и фрагменты хроматограмм семей 1, 2, 3. Верхняя последовательность (хроматограмма – семья 3) соответствует дикому типу, а нижняя соответствует последовательности с новой мутацией.
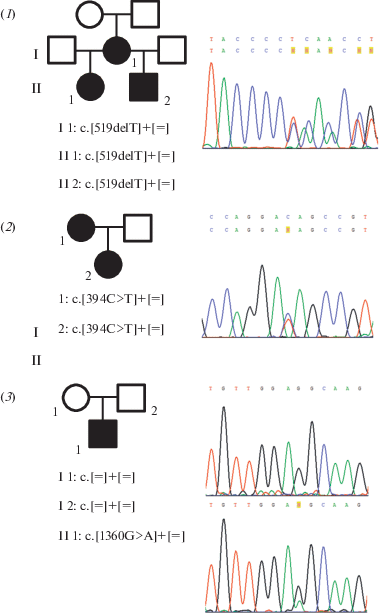
Полученные данные клинического, анамнестического, семейного и молекулярного исследований в совокупности позволяют классифицировать нонсенс-мутацию c.394C>T (p.Gln132*), делеции c.519delT (p.Gln174Asnfs*66) и c.858delC (p.Ile286Leufs*79) и миссенс-мутацию c.1360G>A (p.Gly454Ser) как патогенные варианты и рассматривать все четыре мутации как причину БОР-синдрома согласно практическим рекомендациям [23].
Таким образом, вклад генетической патологии, обусловленной мутациями в гене EYA1, в заболеваемость БОР-синдромом среди исследованных российских пациентов составил 57% (4 из 7 неродственных пробандов), что соответствует зарубежным данным, согласно которым около половины случаев синдрома составляет данная генетическая форма [8]. У остальных трех пробандов, у которых не выявлены патогенные и вероятно-патогенные варианты гена EYA1, возможно наличие протяженных делеций/дупликаций или мутации в регуляторных областях гена EYA1, или мутации в других известных (SIX1 и SIX5) или неизвестных генах БОР-синдрома. В связи с низкой информативностью исследования генов SIX1 и SIX5 у пациентов с БОР-синдромом для данных случаев целесообразно продолжение исследования методами полногеномного или полноэкзомного секвенирования.
Список литературы
Маркова Т.Г. Клинические признаки бранхио-ото-ренального синдрома в семье с положительными результатами исследования гена EYA1 // Вестн. оториноларингологии. 2006. № 6. С. 25–28.
Fraser F.C., Ling D., Clogg D., Nogrady B. Genetic aspects of the BOR syndrome–branchial fistulas, ear pits, hearing loss, and renal anomalies // Am. J. Med. Genet. 1978. № 2. P. 241–252.
Melnick M., Bixler D., Silk K. et al. Autosomal dominant branchiootorenal dysplasia // Birth Defects Orig. Art. Ser. 1975. V. 11. № 5. P. 121–128.
Chang E.H., Menezes M., Meyer N.C. et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences // Hum. Mutat. 2004. № 23. P. 582–589.
Fraser G.R. The Causes of Profound Deafness in Childhood. Baltimore, MD: Johns Hopkins Univ. Press, 1976.
Fraser F.C., Sproule J.R., Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss // Am. J. Med. Genet. 1980. V. 7. № 3. P. 341–349.
Castiglione A., Melchionda S., Carella M. et al. EYA1-relatef disorders: two clinical cases and a literature review // Int. J. Pediatr. Otorhinolaryngol. 2014. V. 78. № 8. P. 1201–1210. doi https://doi.org/10.1016/j.ijporl.2014.03.032
Smith R.J.H. Branchiootorenal spectrum disorders. Gene ReviewsTM (Internet). University of Washington, Seattle, WA, 1999. Mar 19 [Updated 2013] Jun 30; Cited 23 February 2014. Available from URL: http:// www.ncbi.nlm.nih.gov/books/NBK1380/.
Hoskins B.E., Cramer C.H., Silvius D. et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome // Am. J. Hum. Genet. 2007. V. 80. P. 800–804.
Kochhar A., Orten D.J. Sorensen J.L. et al. SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurrent missense mutation associated with BOR // Hum. Mut. 2008. V. 29. P. 565. doi https://doi.org/10.1002/ humu.20714
Abdelhak S., Kalatzis V., Heilig R. et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family // Nature Genet. 1997. V. 15. P. 157–164.
Haan E.A., Hull Y.J., White S. et al. Tricho-rhino-phalangeal and branchio-oto syndromes in a family with an inherited rearrangement of chromosome 8q // Am. J. Med. Genet. 1989. V. 32. P. 490–494.
Ruf R.G., Xu P.X., Silvius D. et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes // Proc. Natl Acad. Sci. USA. 2004. V. 101. P. 8090–8095.
Zhang Y., Knosp B.M., Maconochie M. et al. A comparative study of Eya1 and Eya4 protein function and its implication in branchio-oto-renal syndrome and DFNA10 // J. Assoc. Res. Otolaryngol. 2004. № 5. P. 295–304.
Wawersik S., Maas R.L. Vertebrate eye development as modeled in Drosophila // Hum. Mol. Genet. 2000. V. 9. P. 917–925.
Fortunato S.A., Leininger S., Adamska M. Evolution of the Pax-Six-Eya-Dach network: the calcisponge case study // Evodevo. 2014. V. 5. P. 23. doi https://doi.org/10.1186/2041-9139-5-23
Wayne S., Robertson N.G., DeClau F. et al. Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus // Hum. Mol. Genet. 2001. V. 10. № 3. P. 195–200.
Rebay I., Silver S.J., Tootle T.L. New vision from Eyes absent: transcription factors as enzymes // Trends Genet. 2005. V. 21. № 3. P. 163–171.
Rayapureddi J.P., Kattamuri C., Steinmetz B.D. et al. Eyes absent represents a class of protein tyrosine phosphatases // Nature. 2003. V. 426. P. 295–298.
Tootle T.L., Silver S.J., Davies E.L. et al. The transcription factor Eyes absent is a protein tyrosine phosphatase // Nature. 2003. V. 426. P. 299–302.
Migliosi V., Flex E., Guida V. et al. Identification of five novel BOR mutations in human EYA1 gene associated with branchio-oto-renal syndrome by a DHPLC-based assay // Clin. Genet. 2004. V. 66. P. 478–480. doi https://doi.org/10.1111/j.1399-0004.2004.00318
https://www.thermofisher.com.
Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных, полученных методами массового параллельного секвенирования (MPS) // Мед. генетика. 2017. Т. 16. № 7. С. 4–17.
Bliznetz E.A., Tverskaya S.M., Zinchenko R.A. et al. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: the unique splice site mutation in TCIRG1 gene spread by the founder effect // Eur. J. Hum. Genet. 2009. V. 17. № 5. P. 664–672. doi https://doi.org/10.1038/ejhg.2008.234
Chen A., Francis M., Ni L. et al. Phenotypic manifestations of branchio-oto-renal syndrome // Am. J. Med. Genet. 1995. V. 58. P. 365–370.
Propst E.J., Blaser S., Gordon K.A. et al. Temporal bone findings on computed tomography imaging in branchio-oto-renal syndrome // Laryngoscope. 2005. V. 115. P. 1855–1862.
Song M.H., Kwon T.J., Kim H.R. et al. Mutational analysis of EYA1, SIX1 and SIX5 genes and strategies for management of hearing lossin patients with BOR/BO syndrome // PLoS One. 2013. V. 8. № 6. P. e67236. doi https://doi.org/10.1371/journal.pone.0067236
Toriello H.V., Cohen M.M., Gorlin R.J. et al. Hereditary Hearing Loss and Its Syndromes. Oxford; N.Y., 2004. 502 p.
Unzaki A., Morisada N., Nozu K. et al. Clinically diverse phenotypes and genotypes of patients with branchio-oto-renal syndrome // J. Hum. Genet. 2018. V. 63. № 5. P. 647–656. doi https://doi.org/10.1038/s10038-018-0429-8
Дополнительные материалы отсутствуют.