

Генетика, 2020, T. 56, № 6, стр. 704-713
FISH-диагностика хромосомной транслокации с использованием технологии синтеза локус-специфичных ДНК-зондов на основе ПЦР длинных фрагментов
Д. И. Жигалина 1, *, Н. А. Скрябин 1, О. Ю. Васильева 1, М. Е. Лопаткина 1, С. А. Васильев 1, В. М. Сивоха 1, Е. О. Беляева 1, Р. Р. Савченко 1, Л. П. Назаренко 1, И. Н. Лебедев 1
1 Научно-исследовательский институт медицинской генетики, Томский национальный исследовательский медицинский центр РАН
634050 Томск, Россия
* E-mail: darya.zhigalina@medgenetics.ru
Поступила в редакцию 28.06.2019
После доработки 10.08.2019
Принята к публикации 11.09.2019
Аннотация
Детекция хромосомных транслокаций в субтеломерных участках хромосом представляет собой серьезную диагностическую проблему, поскольку транслокации в этих регионах с участием фрагментов хромосом небольшой протяженности сложно обнаружить при проведении стандартного кариотипирования с помощью методов дифференциального окрашивания. В настоящей работе разработана технология получения ДНК-зонда на уникальные последовательности субтеломерных регионов хромосом на основе ПЦР длинных фрагментов на примере определения родительского происхождения несбалансированной транслокации между хромосомами 5 и 8. По результатам FISH-анализа у пробанда с задержкой физического, моторного, психического и речевого развития и у сибса с задержкой речевого и психического развития была подтверждена выявленная ранее методом хромосомного микроматричного анализа (aCGH) несбалансированная транслокация der(5)t(5;8)(р15.33;q24.22). Размер делетированного участка на хромосоме 5 составил 2.4 млн пн, а протяженность дупликации 8q24.22qter – 11.7 млн пн. С помощью синтезированного локус-специфичного ДНК-зонда в кариотипе матери была обнаружена сбалансированная транслокация 46,XX,t(5;8)(p15.33;q24.22), не выявленная при метафазном анализе G-окрашенных хромосом, при этом у отца сбалансированная транслокация была исключена. Представленный в исследовании метод позволяет на базе молекулярно-цитогенетической лаборатории синтезировать локус-специфичные ДНК-зонды с помощью ПЦР длинных фрагментов для оперативной диагностики криптических хромосомных перестроек.
Диагностика транслокаций в субтеломерных участках представляет собой серьезную методическую проблему. В последние годы появляется все больше доказательств того, что небольшие частичные делеции и дупликации субтеломерных участков хромосом человека происходят чаще, чем предполагалось ранее [1]. Криптические транслокации в этих регионах сложно обнаружить при проведении стандартного кариотипирования с помощью дифференциального окрашивания хромосом. Вероятно, это связано с тем, что небольшие изменения G‑негативных (бледно-окрашенных) участков хромосом, расположенных в большинстве субтеломерных регионов, трудно визуализировать с достаточной точностью. Известно, что в уникальных для каждого субтеломерного региона последовательностях ДНК, находящихся на расстоянии 100–300 тысяч пар нуклеотидов (тпн) от конца хромосом, отмечается высокая концентрация генов и ряда генов-кандидатов для распознаваемых синдромов [1–3]. Эти данные позволяют предположить, что субмикроскопические делеции или дупликации в субтеломерных регионах могут иметь непропорционально бóльшие клинические последствия, чем дисбалансы аналогичного размера в других участках хромосом. При необходимости выявления хромосомных перестроек в субтеломерных регионах использование микроматричной сравнительной геномной гибридизации (aCGH) представляется хорошей альтернативой стандартному цитогенетическому анализу, однако aCGH не позволяет выявлять сбалансированные транслокации [4].
По мере описания микроделеционных синдромов, связанных с субтеломерными регионами, получение локус-специфичных теломерных ДНК-зондов становится все более актуальным для решения конкретных диагностических задач. Детекция несбалансированной транслокации с помощью aCGH у пациента позволяет акцентировать внимание на дальнейшем поиске сбалансированной транслокации для остальных членов семьи с помощью флуоресцентной гибридизации in situ (FISH). Полученные при микроматричном анализе координаты разрывов хромосом дают возможность создать ДНК-зонды, комплементарные целевым последовательностям ДНК, для выявления транслокации. Такие ДНК-библиотеки могут быть созданы благодаря использованию метода ПЦР длинных фрагментов, который дает возможность получать ампликоны размером от 3–5 до 30 тпн [5]. Таким образом, на базе молекулярно-цитогенетической лаборатории могут быть синтезированы ДНК-зонды на целевые последовательности ДНК, которые при проведении FISH будут визуализироваться на метафазных хромосомах. В настоящей работе мы сообщаем об использовании данного алгоритма для диагностики транслокации между хромосомами 5 и 8 в семье пациента с синдромом кошачьего крика.
МАТЕРИАЛЫ И МЕТОДЫ
Пациент У., мальчик (пробанд). Возраст на момент обследования 3 года. В 2–3 года мать обеспокоилась отсутствием речи у ребенка. У пациента отмечается задержка физического, моторного, психического и речевого развития. Пациент был рожден от шестой беременности в результате физиологических родов в 37 нед. Беременность протекала нормально. Вес при рождении составил 2490 г, а рост 51 см. При рождении были обнаружены микрогнатия и расщелина нёба, которые были прооперированы. В связи с чем на момент обращения у пациента не было фенотипических дефектов. Кариотип пациента по данным метафазного анализа G-окрашенных хромосом – 46,XY.
У пациента есть старшая сестра (сибс) с задержкой речевого и психического развития без положительной динамики. Возраст на момент обследования 6 лет. Судороги с двух лет. Наблюдается у невропатолога, основной диагноз: органическое поражение центральной нервной системы. Есть сопутствующие заболевания: гиперметропия средней степени, амблиопия, содружественное сходящееся косоглазие, а также субклинический гипотиреоз. Кариотип сибса – 46,ХХ. Пациент и его сестра были направлены в лабораторию цитогенетики НИИ медицинской генетики Томского НИМЦ для исключения микроделеционных синдромов с помощью микроматричной сравнительной геномной гибридизации. От родителей пациента было получено информированное добровольное согласие на проведение исследования.
ДНК пробанда и сибса была выделена из лимфоцитов периферической крови методом фенол-хлороформной экстракции. Поиск несбалансированных структурных нарушений хромосом осуществляли методом микроматричной сравнительной геномной гибридизации (aCGH) с использованием микрочипа SurePrint G3 Human CGH, 8×60K (Agilent Technologies, США) в соответствии с протоколом производителя. В качестве контрольного образца при анализе ДНК пробанда была использована ДНК индивидов мужского пола с нормальным кариотипом (#5190-4370, Agilent Technologies), а при анализе ДНК сибса – ДНК индивидов женского пола с нормальным кариотипом (#5190-4371, Agilent Technologies). Тестируемая и контрольная ДНК была мечена флуоресцентными красителями Cy5 и Cy3 соответственно. Гибридизацию проводили в течение 16 ч при 67°C. Сканирование микрочипов было проведено с помощью сканера SureScan (Agilent Technologies, США), а анализ результатов гибридизации проводили с использованием программного обеспечения CytoGenomics (v3.0.2.11) (Agilent Technologies). Интерпретация результатов была проведена с использованием онлайн‑версии Каталога наследственных болезней “Менделевское наследование у человека” (OMIM) [6].
Для выявления транслокации между хромосомами 5 и 8 с помощью FISH нами был создан ДНК-зонд, специфичный к субтеломерному региону длинного плеча хромосомы 8q24.22qter. Выбор региона, на который создавался ДНК-зонд, основывался на возможности эффективной детекции как сбалансированной, так и несбалансированной транслокации при анализе метафазных хромосом пациента и членов его семьи. Использование локус-специфичного ДНК-зонда на субтеломерный регион хромосомы 5p не позволило бы точно диагностировать несбалансированную транслокацию, так как по результатам гибридизации теоретически должен быть виден только один флуоресцентный сигнал (рис. 1). В этом случае не может быть полностью исключена возможность получения ложно-положительного результата из-за низкой интенсивности второго флуоресцентного сигнала либо из-за наложения флуоресцентных сигналов. При использовании локус-специфичного ДНК-зонда на субтеломерный регион хромосомы 8q на метафазных хромосомах будут выявлены три флуоресцентных сигнала, что позволит получить достоверные результаты. Таким образом, нами были подобраны праймеры, позволяющие амплифицировать несколько тандемно расположенных фрагментов на длинном плече хромосомы 8 (табл. 1).
Рис. 1.
Теоретически ожидаемые результаты FISH при использовании центромеро-специфичного ДНК-зонда на хромосому 8 (красная метка) и локус-специфичного ДНК-зонда на субтеломерный регион хромосомы 5 (зеленая метка), локус-специфичного ДНК-зонда на субтеломерный регион хромосомы 8 (зеленая метка).
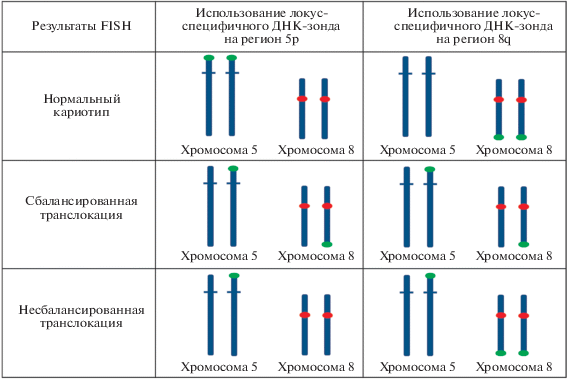
Таблица 1.
Праймеры для амплификации участка ДНК на длинном плече хромосомы 8
| № | Название | Последовательность | Tm, °С | Длина продукта, пн |
|---|---|---|---|---|
| 1 | 8q subtel_LR1-F | 5'-ATAGAGGCTTGGACCCGACT-3' | 60.0 | 9276 |
| 8q subtel_LR1-R | 5'-GCTGGCCCACAAGAGAGATT-3' | 60.0 | ||
| 2 | 8q subtel_LR2-F | 5'-AGAAGGGAGCATAGACCCGT-3' | 60.0 | 6240 |
| 8q subtel_LR2-R | 5'-TCTAGGCACTCCTGGACCTC-3' | 60.0 | ||
| 3 | 8q subtel_LR3-F | 5'-GAAGGCGGTGTTGTGTTTCC-3' | 59.9 | 6883 |
| 8q subtel_LR3-R | 5'-GCAGTCCTCTTGACATGCCT-3' | 60.0 | ||
| 4 | 8q subtel_LR4-F | 5'-ACAGCACGCTAAGTGCTTCT-3' | 59.9 | 6305 |
| 8q subtel_LR4-R | 5'-GTACTCGCCTGATGGAGAGC-3' | 59.9 | ||
| 5 | 8q subtel_LR4-F | 5'-ACAGCACGCTAAGTGCTTCT-3' | 59.9 | 9111 |
| 8q subtel_LR4-R | 5'-GAGGGCCCCCTAGGAAGTTA-3' | 60.0 | ||
| 6 | 8q subtel_LR5-F | 5'-GAGGCCCTCACACAAGTACC-3' | 60.0 | 5880 |
| 8q subtel_LR5-R | 5'-GTTGGACGTCAGAATCCCGT-3' | 60.0 | ||
| Суммарная длина продукта | 43 695 | |||
Суммарная длина фрагментов должна была превышать 30 тпн, чтобы ДНК-зонд был хорошо визуализирован на метафазных хромосомах при проведении FISH-анализа. Получаемые фрагменты должны быть расположены максимально близко на хромосоме, но не перекрываться. На каждые 10 тпн подбирали одну пару праймеров. В настоящей работе для получения последовательности нуклеотидов в регионе частичной трисомии 8q24.22qter нами был использован геномный браузер UCSC (University of California, Санта Круз), который содержит информацию о последовательностях геномов [7]. В дальнейшем полученную последовательность нуклеотидов использовали для подбора праймеров с помощью стандартных биоинформатических программ (BLAST), которые предоставляет Национальный центр биотехнологической информации США (англ. National Center for Biotechnological Information, NCBI) [8]. Праймеры подбирали с учетом следующих условий:
1. Все праймеры должны иметь близкую температуру плавления (Tm) и температуру отжига (±1°С).
2. Длина праймера может варьировать от 15 до 30 пн.
3. Специфичность праймеров должна быть не менее 70%.
4. Внутренняя гомология не должна превышать 3 пн.
5. Содержание GC-динуклеотидов может варьировать в диапазоне от 45 до 55%.
Нами были исключены праймеры с полипиримидиновыми или полипуриновыми протяженными участками, а также праймеры и сайты-мишени, температура плавления вторичной структуры которых была выше либо эквивалентна температуре плавления самого праймера. Кроме того, нами оценивалась возможность гомо- и гетеродимеризации праймеров, так как это может привести к снижению выхода конечного продукта ПЦР.
Наработку ДНК-продуктов проводили с помощью ПЦР длинных фрагментов с использованием набора БиоМастер LR HS-ПЦР-Color (2×) (#MHC040-100, Biolabmix, Россия). Наработку фрагментов проводили на амплификаторе SureCycler 8800 (Agilent Technologies) при следующем режиме: 94°С, 4 мин; 10 циклов: 94°С, 20 с; 57°С, 30 с; 68°С, 13 мин; 20 циклов: 94°С, 20 с; 57°С, 30 с; 68°С, 13 мин (+10 с за цикл к этапу элонгации). При проведении электрофореза в агарозном геле для всех амплифицированных фрагментов были обнаружены соответствующие бэнды.
Для идентификации хромосомы 8 нами была получена центромеро-специфичная ДНК-библиотека на регион 8p11.1-q11. Для этого использовались трансформированные клоны E. coli с плазмидами, содержащими вставки центромеро-специфичных фрагментов α-сателлитной ДНК. Процедуру по выделению плазмидной ДНК со вставками центромеро-специфичных фрагментов ДНК хромосом человека из бактериальных клеток E. сoli проводили с помощью набора Wizard Minipreps (# BC021, Евроген, Россия).
С помощью реакции ник-трансляции было проведено мечение центромеро-специфичной и локус-специфичной ДНК-библиотек с использованием флуоресцентных красителей fluorescein-12-dUTP и TAMRA-5-dUTP соответственно. Гибридизацию ДНК-зондов на препараты метафазных хромосом проводили в гибридизационной камере ThermoBrite (Abbott Molecular, США) в течение 24 ч при 37°С с 50-кратным избытком C0t-1 ДНК (СибХромоТех, Россия). Для детекции гибридизационных сигналов был использован люминесцентный микроскоп AxioImager.Z2 (Сarl Zeiss, Германия). Результаты FISH были проанализированы с помощью программного продукта Isis (Metasystems, Германия). Исследование выполнено с использованием оборудования Центра коллективного пользования “Медицинская геномика” Томского НИМЦ и ресурсов биологической коллекции “Биобанк населения Северной Евразии” НИИ медицинской генетики Томского НИМЦ.
РЕЗУЛЬТАТЫ
Анализ с помощью микроматричной сравнительной геномной гибридизации позволил выявить идентичные несбалансированные транслокации у пробанда и у сибса соответственно:
46,XY,der(5)t(5;8)(p15.3;q24)mat.arr[hg19]5р15.33(204737_2574299)×1,8q24.22.q24.3(134557660_146250824)×3 и
46,XХ,der(5)t(5;8)(p15.3;q24)mat.arr[hg19]5р15.33(204737_2574299)×1,8q24.22.q24.3(34557660_146250824)×3
(рис. 2). В регионе делеции 5p15.33, протяженность которого составила 2.4 млн пн, локализовано 43 гена, при этом мутации в семи из них (SDHA, SLC6A19, TERT, SLC6A3, NDUFS6, TRIP13, SLC9A3) являются патогенетически значимыми в медицинской онлайн базе данных “Менделевское наследование у человека” (OMIM) [6]. Делеции гена TERT могут быть причиной возникновения ряда фенотипических признаков при синдроме кошачьего крика (Cri-du-chat syndrome, OMIM 123450). Вариации в числе копий тандемных повторов в 3'-нетранслируемой области гена SLC6A3 ассоциированы с развитием идиопатической эпилепсии, синдрома дефицита внимания и гиперактивности (OMIM 143465). Мутации в данном гене также связаны с развитием инфантильного паркинсонизма-дистонии, тип I (OMIM 613135) и недостаточности комплекса I дыхательной цепи митохондрий (OMIM 618232), состояния, приводящего к широкому спектру клинических проявлений, включая заболевания новорожденных и нейродегенеративные расстройства у взрослых.
Рис. 2.
Гибридизационные профили хромосом 5 и 8 у пробанда (а) и сибса (б) по результатам микроматричной сравнительной геномной гибридизации.
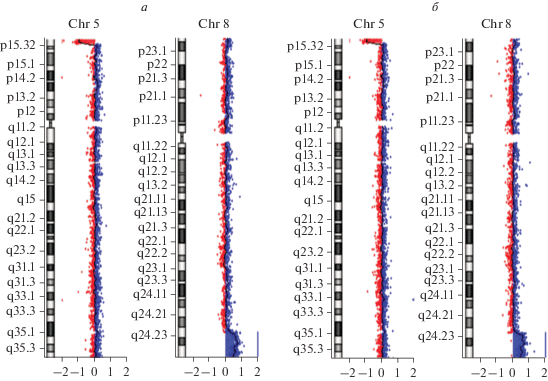
В регионе дупликации 8q24.22-q24 протяженностью 11.7 млн пн, локализовано 154 гена, при этом мутации в 17 из них (ZFAT, KCNK9, TRAPPC9, SLURP1, CYP11B1, CYP11B2, GRIHBP1, FAM83H, PUF60, PLEC, OPLAH, GPAA1, CYC1, DGAT1, SLC52A2, SLC39A4, RECQL4) являются патогенетически значимыми, согласно базе данных OMIM. Мутации в гене KCNK9, импринтированном в мозге, ассоциированы с развитием синдрома Бирк-Барела, характеризующегося умственной отсталостью (Birk-Barel mental retardation dysmorphism syndrome, OMIM 612292). Перестройки, затрагивающие ген TRAPPC9, также связаны с развитием умственной отсталости (OMIM 613192). Мутации в гене SLC52A2, ответственном за синтез транспортера рибофлавина, ассоциированы с развитием синдрома Браун-Виалетто-Ван Лир (Brown-Vialetto-Van Laere syndrome 2, OMIM 614707). Bonaglia с коллегами [9] полагают, что дупликация гена GRINA (OMIM 138251), расположенного в регионе 8q24 (OMIM 600669) и ответственного за синтез глутамат-связывающей субъединицы NMDA-рецептора ионного канала, может быть связана с формированием генерализованной идиопатической эпилепсии.
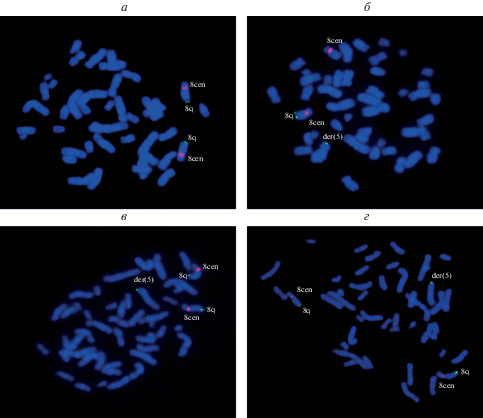
FISH с использованием центромеро-специфичного и полученного нами локус-специфичного ДНК-зонда на хромосому 8 дала возможность проанализировать метафазные хромосомы пациентов, а также их отца и матери на наличие транслокации между хромосомами 5 и 8. У отца обе флуоресцентные метки были обнаружены на хромосоме 8 в соответствующих нормальному расположению регионах, на основании чего транслокация у него была исключена (рис. 3,а). У матери была выявлена сбалансированная транслокация. Флуоресцентная метка, соответствующая субтеломерному региону 8q, отсутствовала на одной из хромосом 8 и была обнаружена на коротком плече хромосомы 5 (рис. 3,б). Отсутствие фенотипических проявлений транслокации у матери связано с тем, что данная транслокация является сбалансированной и не влечет за собой изменений в транскрипции генов. У пробанда и сибса несбалансированная транслокация, выявленная с помощью aCGH, также была подтверждена и методом FISH. При проведении FISH флуоресцентные метки на хромосомах 8 были расположены в соответствующих регионах, однако на коротком плече одной хромосомы 5 также произошла гибридизация ДНК-зонда, специфичного к длинному плечу хромосомы 8. В результате нами были выявлены три флуоресцентных сигнала, соответствующих субтеломерному региону хромосомы 8 – частичная трисомия 8q24.22qter (рис. 3,в, г).
Рис. 3.
FISH центромеро-специфичного ДНК-зонда на хромосому 8, меченного TAMRA-5-dUTP, и локус-специфичного ДНК-зонда на субтеломерный регион длинного плеча хромосомы 8, меченного fluorescein-12-dUTP. а – метафазные хромосомы отца; б – метафазные хромосомы матери; в – метафазные хромосомы пробанда; г – метафазные хромосомы сибса. 8q – флуоресцентный сигнал в субтеломерном регионе длинного плеча хромосомы 8; 8сen – флуоресцентный сигнал в области центромеры хромосомы 8; der(5) – флуоресцентный сигнал в субтеломерном регионе короткого плеча хромосомы 5.
Умственная отсталость является распространенной патологией (2–3% от общей численности детей), причины которой более чем в 50% случаев остаются невыясненными. Субмикроскопические хромосомные аномалии в 4–28% случаев являются причиной умственной отсталости [3]. В настоящем исследовании фенотип пациента и сибса обусловлен делецией в дистальном участке короткого плеча хромосомы 5 и увеличением копийности субтеломерного участка длинного плеча хромосомы 8. Известно, что делеция короткого плеча хромосомы 5 приводит к формированию синдрома кошачьего крика, который характеризуется умственной отсталостью и задержкой развития речевых и физических навыков [10]. Cиндром кошачьего крика может формироваться при делеции всего короткого плеча хромосомы 5 или только региона 5р15 [11]. Размер делеции может варьировать от 5 до 40 млн пн [12]. В работе Mainardi с коллегами [13] была подтверждена связь между наличием делеции в регионе р15.3 и формированием синдрома кошачьего крика, а также задержкой речи. Другая исследовательская группа сообщила о ребенке с синдромом кошачьего крика и делецией 5р14.3, унаследованной от матери [14]. Zhang с соавт. [15] использовали сравнительную геномную гибридизацию для анализа 94 пациентов с синдромом кошачьего крика, которые были тщательно оценены на наличие характерного детского плача, задержки речи, лицевых дисморфий и умственной отсталости. У большинства пациентов выявлены делеции, включающие 5p (84%, 79/94). У 67 пациентов была детектирована терминальная делеция, а у 12 – интерстициальная. Medina с коллегами [16] определили, что ген CTNND2 (OMIM 604275), находящийся в регионе 5p15.2, вовлечен в формирование фенотипа умственной отсталости и синдрома кошачьего крика. В проведенном ими исследовании была обнаружена сильная корреляция между гемизиготной потерей CTNND2 и тяжелой умственной отсталостью. Авторы пришли к выводу, что эти результаты и функция CTNND2 как нейрон-специфического белка, экспрессируемого на ранних стадиях развития и участвующего в подвижности клеток подтверждают роль гена CTNND2 в умственной отсталости при синдроме кошачьего крика в случаях гаплонедостаточности, когда данный ген присутствует только в одной копии.
Дупликация субтеломерного участка длинного плеча хромосомы 8 является редкой хромосомной аномалией. Симптомы и степень тяжести фенотипических нарушений зависят от размера и местоположения дупликации, от того, какие гены участвуют, и от наличия других хромосомных аномалий. В целом для пациентов с частичной трисомией 8q характерна задержка роста, плоский затылок с низкой задней линией волос, низко посаженные и маленькие уши, гипертелоризм, густые брови, округлая колумелла и широкие ноздри, опущенные углы рта и малой нижней челюсти, короткая шея, клино- и камптодактилия [17]. Врожденные аномалии встречаются редко. Тем не менее у одной пациентки был зафиксирован атриовентрикулярный дефект межжелудочковой перегородки, у ее сестры – синдром лобной доли [18]. У мальчика трех лет был также выявлен дефект межжелудочковой перегородки, а у двух пациентов наблюдались другие дефекты сердца [19]. Один пациент в возрасте 15 лет имел тяжелую степень умственной отсталости, в то время как у остальных умственная отсталость была умеренной [18]. Phelan с соавт. [20] также отметили наличие умственной отсталости, проявляющейся в разной степени у членов семьи с частичной трисомией 8q24.1qter. В 2009 г. был описан случай несбалансированной транслокации der(3)t(3;8)(p25;q24) у девочки 9 лет с тяжелой степенью умственной отсталости, с задержкой внутриутробного и постнатального развития, микроцефалией, капиллярными гемангиомами лба и пероральной области, синофризом, птозом, микрогнатией, деформированными ушами, клинодактилией, гипотонией, двигательной отсталостью [21].
В литературе представлен случай частичной трисомии 8q de novo [22], хотя наиболее часто данная хромосомная аберрация наследуется от родителей вследствие мейотической сегрегации сбалансированных транслокаций. Так, в одном из описаний у двух сестер аберрация 8q была унаследована от матери с транслокацией [18], а еще у одной девочки ее формирование стало следствием перицентрической инверсии у матери [23]. Kausch с соавт. был описан фенотип шестнадцатилетней девушки с дупликацией региона 8q24.2qter и региона 15q14pter, унаследованных от матери-носительницы сбалансированной транслокации [24]. В исследовании Kleczkowska с коллегами у пяти из шести пациентов формирование частичной трисомии 8q также было связано с наличием семейной транслокации [22].
Тем не менее достоверная оценка вклада частичной трисомии 8q24qter в фенотип наших пациентов затруднена в связи с небольшим числом описанных в литературе клинических случаев. Кроме того, как в представленном нами случае, так и в описанных в литературе помимо частичной трисомии 8q24qter пациенты являются носителями других несбалансированных хромосомных аберраций, вклад которых трудно дифференцировать [18, 21, 24–27].
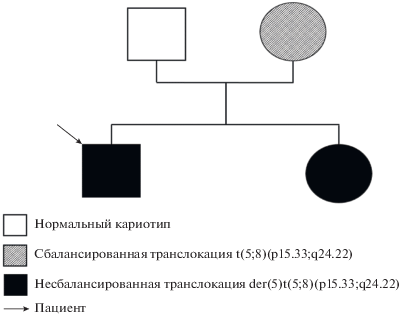
На основе полученных с помощью методов aCGH и FISH результатов нам удалось проследить наследование транслокации в семье пациента У. (рис. 4). Было показано, что диагностированная у пробанда и сибса несбалансированная транслокация der(5)t(5;8)(р15.33;q24.22) имеет материнское происхождение.
ОБСУЖДЕНИЕ
В данной работе представлены результаты применения технологии создания и использования ДНК-зонда на уникальные последовательности субтеломерного региона хромосомы 8 для определения родительского происхождения несбалансированной транслокации der(5)t(5;8)(р15.33;q24.22). Анализ ДНК пациента и сибса методом микроматричной сравнительной геномной гибридизации позволил точно определить координаты разрывов хромосом. На основе полученных координат в дальнейшем был проведен FISH-анализ с использованием полученного с помощью ПЦР длинных фрагментов локус-специфичного ДНК-зонда. В результате у матери была выявлена сбалансированная транслокация 46,XX,t(5;8)(p15.33;q24.22), в то время как у отца данная транслокация была исключена.
При создании локус-специфичного ДНК‑зонда наиболее существенным является вопрос о регионе, на который должен будет гибридизоваться полученный зонд. Для исключения возможности получения ложно-положительных или ложно-отрицательных результатов следует выбирать регион, который будет дуплицирован у пациента с несбалансированной транслокацией. В таком случае на метафазных хромосомах будут детектированы три флуоресцентных сигнала, соответствующих региону с частичной трисомией. На основании выбранного региона проводится подбор праймеров.
Для хорошей визуализации флуоресцентного сигнала на метафазных хромосомах размер ДНК-зонда должен превышать 30 тпн. ПЦР длинных фрагментов с помощью набора БиоМастер LR HS-ПЦР-Color (2×) (#MHC040-100, Biolabmix, Россия) позволяет амплифицировать фрагменты размером 15 тпн. В состав набора входят две разные ДНК-полимеразы. HS-Taq ДНК-полимераза представляет собой смесь рекомбинантного фермента Taq ДНК-полимеразы и моноклональных антител. Наличие “горячего старта” существенно повышает чувствительность и специфичность реакции по сравнению с Taq-полимеразой. Pfu ДНК-полимераза – это термостабильная и гиперактивная мономерная полимераза, которая значительно повышает точность синтеза ДНК по сравнению с Taq-полимеразой. Тем не менее с целью повышения эффективности ПЦР в настоящей работе в среднем на каждые 10 тпн подбиралась одна пара праймеров (прямой и обратный). Таким образом, для получения ДНК-зонда требуемой длины необходимо, чтобы были подобраны праймеры минимум на три близко расположенных участка ДНК. Для удобства проведения ПЦР все подобранные праймеры должны иметь близкую температуру плавления.
Для подбора праймеров используются стандартные биоинформатические программы (BLAST) [8]. Данная задача может быть выполнена в течение одного рабочего дня. Постановка ПЦР длинных фрагментов с использованием подобранных праймеров также занимает один день. Качество полученных ДНК-библиотек оценивается с помощью электрофореза в агарозном геле по наличию бэнда соответствующей длины, а также по результатам измерения концентрации ДНК. После чего производится мечение полученной ДНК-библиотеки флуоресцентным красителем в ходе реакции ник-трансляции. Для постановки флуоресцентной гибридизации in situ с использованием полученных локус-специфичных ДНК-зондов необходимо два рабочих дня. Кроме того, для идентификации хромосомы, на которую был создан ДНК-зонд, необходимо дополнительно использовать центромеро-специфичный ДНК-зонд, меченный другим флуоресцентным красителем относительно локус-специфичного ДНК-зонда. В таком случае по результатам FISH-анализа появится возможность оценить специфичность гибридизации полученного ДНК-зонда, а также дифференцировать нормальный кариотип, сбалансированную и несбалансированную транслокацию. Таким образом, представленный метод позволяет на базе молекулярно-цитогенетической лаборатории создавать ДНК-зонды на уникальные последовательности хромосом с помощью ПЦР длинных фрагментов для оперативной диагностики криптических хромосомных перестроек.
Исследование поддержано грантом Федерального фонда содействия развитию малых форм предприятий в научно-технической сфере в рамках программы “УМНИК” на проведение НИР по договору № 10661ГУ/19966 от 24.10.2016.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Martin C.L., Ledbetter D.H. Molecular cytogenetic analysis of telomere rearrangements // Curr. Protoc. Hum. Genet. 2015. V. 84. № 1. P. 8–11. https://doi.org/10.1002/0471142905.hg0811s84
Saccone S., De Sario A., Della-Valle G. et al. The highest gene concentrations in the human genome are in telomeric bands of metaphase chromosomes // Proc. Natl Acad. Sci. USA. 1992. V. 89. № 11. P. 4913–4917. https://doi.org/10.1073/pnas.89.11.4913
De Vries B.B.A., Winter R., Schinzel A. et al. Telomeres: a diagnosis at the end of the chromosomes // J. Med. Genet. 2003. V. 40. № 6. P. 385–398. https://doi.org/10.1136/jmg.40.6.385
Shinawi M., Cheung S.W. The array CGH and its clinical applications // Drug Discovery Today. 2008. V. 13. № 17–18. P. 760–770. https://doi.org/10.1016/j.drudis.2008.06.007
Jia H., Guo Y., Zhao W. et al. Long-range PCR in next-generation sequencing: comparison of six enzymes and evaluation on the MiSeq sequencer // Sci. Rep. 2014. V. 4. P. 5737. https://doi.org/10.1038/srep05737
https://www.omim.org – Онлайн база наследственных болезней “Менделевское наследование у человека” (OMIM).
http://genome.ucsc.edu – геномный браузер UCSC (Univ. California, Santa Cruz).
https://www.ncbi.nlm.nih.gov – Национальный центр биотехнологической информации США (National Center for Biotechnological Information, NCBI).
Bonaglia M.C., Giorda R., Tenconi R. et al. A 2.3 Mb duplication of chromosome 8q24.3 associated with severe mental retardation and epilepsy detected by standard karyotype // Eur. J. Hum. Genet. 2005. V. 13. № 5. P. 586–591. https://doi.org/10.1038/sj.ejhg.5201369
Lejeune J., Lafourcade J., Berger R. et al. Trois cas de deletion partielle du bras court d’un chromosome 5 // CR Acad. Sci. (D). 1963. № 257. P. 3098–3102.
Overhauser J., Huang X., Gersh M. et al. Molecular and phenotypic mapping of the short arm of chromosome 5: sublocalization of the critical region for the cri-du-chat syndrome // Hum. Mol. Genet. 1994. V. 3. № 2. P. 247–252. https://doi.org/10.1093/hmg/3.2.247
Simmons A.D., Goodard S.A., Gallardo T.D. et al. Five novel genes from the cri-du-chat critical region isolated by direct selection // Hum. Mol. Genet. 1995. V. 4. № 2. P. 295–302. https://doi.org/10.1093/hmg/4.2.295
Mainardi P.C., Perfumo C., Cali A. et al. Clinical and molecular characterisation of 80 patients with 5p deletion: genotype–phenotype correlation // J. Med. Genet. 2001. V. 38. № 3. P. 151–158.https://doi.org/10.1136/jmg.38.3.151
South S.T., Swensen J.J., Maxwell T. et al. A new genomic mechanism leading to cri-du-chat syndrome // Am. J. Med. Genet. 2006. V. 140. № 24. P. 2714–2720. https://doi.org/10.1002/ajmg.a.31496
Zhang X., Snijders A., Segraves R. et al. High-resolution mapping of genotype–phenotype relationships in cri du chat syndrome using array comparative genomic hybridization // Am. J. Hum. Genet. 2005. V. 76. № 2. P. 312–326.
Medina M., Marinescu R.C., Overhauser J. et al. Hemizygosity of δ-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome // Genomics. 2000. V. 63. № 2. P. 157–164. https://doi.org/10.1006/geno.1999.6090
Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. N.Y.: de Gruyter, 2001. 982 p.
Schinzel A. Partial trisomy 8q in half-sisters with distinct dysmorphic patterns not similar to the trisomy 8 mosaicism syndrome // Hum. Genet. 1977. V. 37. № 1. P. 17–26.
Romain D.R., Bloxham R.A., Columbano-Green L.M. et al. Familial distal trisomy 8 (q24.13 → qter) // J. Med. Genet. 1989. V. 26. № 2. P. 133–138.
Phelan M.C., Skinner S.A., Cooley L. et al. Familial translocation (8;9) resulting in trisomy 8q24.1 → qter in three generations // Proc. Greenwood Genet. Centar. 1992. № 11. P. 22–26.
Balci S., Aypar E., Beksaç M.S. et al. A severely mentally and motor retarded girl with monosomy 3pter → p25 and trisomy 8q24→qter due to a familial reciprocal translocation t(3;8)(p25;q24) // Genet. Couns. 2009. V. 20. № 2. P. 125–132.
Kleczkowska A., Fryns J.P., Decock P. Inverted distal duplication of the long arm of chromosome 8: borderline intelligence and discrete dysmorphic syndrome // Genet. Couns. 1991. V. 2. № 2. P. 109–113.
Barnes I.C., Kumar D., Bell R.J. A child with a recombinant of chromosome 8 inherited from her carrier mother // J. Med. Genet. 1985. V. 22. № 1. P. 67–70. https://doi.org/10.1136/jmg.22.1.67
Kausch K., Haaf T., Schmid M. Duplication 8q24.2 → → qter and 15q14 → pter resulting from a 3:1 meiotic segregation of a maternal reciprocal translocation // Am. J. Med. Genet. 1988. V. 31. № 4. P. 981–985. https://doi.org/10.1002/ajmg.1320310433
Lejeune J., Rethore M.O. Trisomies of chromosome No 8 // Nobel Symp. 1973. V. 23. P. 214–216.
Sanchez O., Yunis J.J. Partial trisomy 8 (8q24) and the trisomy 8 syndrome // Humangenetik. 1974. V. 23. № 4. P. 297–303.
Fujimoto A., Wilson M.G., Towner J.W. Familial inversion of chromosome 8. An affected child and a carrier fetus // Humangenetik. 1975. V. 27. № 1. P. 67–73.
Дополнительные материалы отсутствуют.