

Генетика, 2021, T. 57, № 5, стр. 516-527
Гены-модификаторы как причина клинического полиморфизма болезни Вильсона–Коновалова
А. Е. Постригань 1, *, И. Ж. Жалсанова 1, Е. А. Фонова 1, Н. А. Скрябин 1
1 Научно-исследовательский институт медицинской генетики, Томский национальный исследовательский медицинский центр Российской академии наук
634050 Томск, Россия
* E-mail: postrigan.anna@medgenetics.ru
Поступила в редакцию 24.06.2020
После доработки 20.10.2020
Принята к публикации 03.11.2020
Аннотация
Более чем за 100 лет после выделения болезни Вильсона–Коновалова в самостоятельную нозологическую форму этиология и патогенез этого заболевания изучены с разных точек зрения – как врачами-клиницистами, так и генетиками. Однако и по нынешний день появляются все новые сведения не только о мутациях в самом гене ATP7B, но и о модифицирующих факторах, влияющих на клиническую картину заболевания. С генетической точки зрения влияние генов-модификаторов неоспоримо, ведь за счет их действия (протективного или компенсирующего) можно объяснить клиническую вариабельность болезни Вильсона–Коновалова и возможно в дальнейшем использовать эти знания в рамках персонализированного лечения пациента. В настоящем обзоре рассматривается влияние различных модификаторов (внутри- и внегенных) на течение и проявление клинических форм болезни Вильсона–Коновалова. Авторами обозначены возможные механизмы возникновения более мягких или более тяжелых форм болезни Вильсона–Коновалова при участии определенных генов.
Болезнь Вильсона–Коновалова (OMIM 277 900, гепатолентикулярная дегенерация, гепатоцеребральная дистрофия, БВК) – тяжелое прогрессирующее наследственное заболевание, в основе которого лежит нарушение экскреции меди из организма, приводящее к избыточному накоплению этого микроэлемента в тканях и сочетанному поражению паренхиматозных органов (прежде всего печени) и головного мозга (преимущественно подкорковых ядер) [1]. Тип передачи этого заболевания аутосомно-рецессивный, проявляется у гомозиготных носителей мутации или у компаунд-гетерозигот [2].
Болезнь Вильсона–Коновалова относится к редким заболеваниям. Распространенность БВК по данным Orphanet составляет 1–9 случаев на 100 000 населения (в среднем 1 на 25 000), ежегодная частота выявления новых случаев – от 1 на 30 000–100 000 населения.
Носителем дефектного гена по ориентировочным оценкам является каждый 90–100-й человек (1%). Еще недавно считалось, что БВК крайне редкое наследственное заболевание, однако проведенные в Европе исследования 2013 г., основанные на анализе результатов секвенирования 2000 экзомов, показали, что гетерозиготное носительство патогенетически значимых мутаций значительно превышает прежние показатели. По последним данным в некоторых странах Европы расчетная частота болезни Вильсона–Коновалова равна 1 : 7000–1 : 9000, тогда как по клиническим данным распространение БВК в этих странах колеблется от 1.2 до 2 : 100 000 [3, 4]. Для РФ информация о частоте встречаемости БВК отсутствует, что связано в первую очередь с этногеографическими особенностями страны. Однако уже появились отдельные исследования расчетной частоты (1 : 10 000) носительства болезни Вильсона–Коновалова в Российской Федерации [5].
Причиной болезни Вильсона-Коновалова являются мутации в гене ATP7B, расположенном в длинном плече 13-й хромосомы (13q14.3). Ген кодирует металлопереносящую аденозинтрифосфатазу (АТФазу) P-типа, которая транспортирует медь в желчь и включает ее в церулоплазмин (гликопротеин, содержит около 95% общего количества меди сыворотки крови человека) [6].
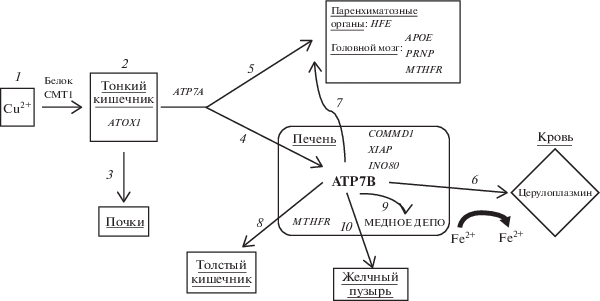
В норме всасывание меди в организме происходит в тонком кишечнике, с помощью транспортного белка CMT1 (Copper Membrane Transporter 1) (рис. 1). CMT1 перемещает медь внутрь клеток, где часть ее связывается с металлотионеином, а оставшаяся – перемещается в комплекс Гольджи с помощью транспортного белка ATOX1. В ответ на повышение концентрации меди в комплексе Гольджи активируется фермент ATP7A и высвобождает медь через воротную вену в печень. В гепатоцитах происходит связывание меди с церулоплазмином и высвобождение комплекса в кровь с помощью белка ATP7B. Кроме того, ATP7B осуществляет экстракцию избытка меди в желчь [7]. Мутации в гене ATP7B приводят к снижению функциональности фермента, в результате чего измененная АТФаза теряет способность транспортировать медь к церулоплазмину и лизосомам [6, 8]. Нарушение транспорта меди приводит к снижению экстракции меди в желчь, накоплению меди в печени, повреждению гепатоцитов и выходу свободной токсичной меди в кровь, с дальнейшим накоплением ее в других тканевых депо – глазах, суставах, почках, головном мозге [6, 8, 9]. Регуляция процесса экстракции меди из гепатоцитов в желчные канальцы осуществляется двумя генами – ATP7B и COMMD1. Избыток меди в печени влечет за собой разрушение клеток за счет окислительного стресса, что в свою очередь приводит к воспалению печени, ее фиброзу и как следствие – к циррозу. Экстракция меди в сыворотку крови осуществляется путем внедрения ионов меди в церулоплазмин (этот процесс также контролируется геном ATP7B). Церулоплазмин является главным медьсодержащим белком в организме (в нем содержится до 95% меди в плазме крови); кроме того, он принимает участие в метаболизме железа, катализируя окисление Fe2+ в Fe3+. Таким образом, метаболические пути меди и железа в организме тесно пересекаются и оказывают влияние друг на друга. Так, при нарушении абсорбции железа в результате мутации в гене HFE происходит его накопление в клетках паренхиматозных органов и усиление их окислительного повреждения. Также деструктивным процессам в клетках способствует повышение уровня гомоцистеина, так как гомоцистеин и медь обладают синергической цитотоксичностью. Уровень гомоцистеина зависит от активности метилфолатредуктазы, которая, в свою очередь, контролируется геном MTHFR. Однако интенсивность повреждения может быть снижена за счет протективного действия гена INO80, стабилизирующего ДНК клеток путем увеличения подвижности хроматина. Накопление меди в других органах вызывает нарушение их функции и снижение качества жизни пациентов. Так, накопление меди в головном мозге приводит к нейродегенеративным процессам, на степень выраженности которых оказывают влияние мутации не только в гене ATP7B, но и в ряде других генов; в частности, продукты генов APOE и PRNP оказывают влияние на процессы миелинизации нервных волокон в головном мозге, что напрямую воздействует на неврологическую компоненту БВК.
Рис. 1.
Патогенез болезни Вильсона–Коновалова. 1 – поступление меди с пищей; 2 – всасывание меди в тонком кишечнике; 3 – выведение меди через почки; 4 – поступление меди в печень через воротную вену; 5 – снабжение медью других органов и систем; 6 – катализ окисления Fe2+ в Fe3+ и выход меди в составе церулоплазмина в кровь; 7 – поступление ионов меди из печени в другие органы; 8 – выведение меди через толстый кишечник; 9 – накопление меди в печени в “медном депо”; 10 – экстракция избытка меди в желчь.
В настоящее время используется классификация, построенная на клинических признаках болезни, сочетания поражения печени и центральной нервной системы, предложенная J. Walsh (1983): бессимптомная форма, печеночная форма, церебральная форма, смешанная форма болезни Вильсона–Коновалова.
Кроме того, согласно классификации Н.В. Коновалова (1960), в зависимости от вовлечения в патологический процесс печени и центральной нервной системы и характера экстрапирамидной симптоматики распознают пять форм болезни Вильсона–Коновалова: брюшная (абдоминальная), ригидно-аритмогиперкинетическая, дрожательно-ригидная, дрожательная и экстрапирамидно-корковая.
При постановке диагноза выраженный клинический полиморфизм признаков болезни Вильсона–Коновалова и их экспрессивность могут затруднять работу врача.
Болезнь Вильсона–Коновалова обладает широким спектром клинических проявлений, зависящих и от конкретного типа мутаций. Большинство миссенс-мутаций приводят к снижению активности фермента (в результате потери целостности белка, мисфолдинга, нарушения взаимодействия между белками, нарушения фосфорилирования, изменения способности связывать АТФ) [10]. Мутации со сдвигом рамки считывания (фрэймшифт-мутации), нонсенс-мутации, инсерции/делеции и мутации сайта сплайсинга приводят к прерыванию (нарушению) синтеза белка АТР7В, что ведет к ранней манифестации заболевания, более низкому уровню церулоплазмина, фульминантной печеночной недостаточности и другим тяжелым клиническим проявлениям БВК, по сравнению с миссенс-мутациями [11, 12].
Интерес к этиологической причине болезни Вильсона–Коновалова не угасает и по сей день. Ряд исследователей из Университета Альберты даже создали открытую базу данных по болезни Вильсона–Коновалова, которая представляет собой сборник научных публикаций и отдельных материалов по исследованиям патологических вариантов мутаций в гене АТР7В [http://www.wilsondisease.med.ualberta.ca/]. Однако данных о специфических мутациях для российской популяции там крайне мало и, кроме того, данная база данных не пополнялась с 2010 г. В настоящее время в гене АТР7В идентифицировано более 800 различных мутаций, а нарушение обмена меди может происходить как на уровне АТФазы, так и на любом уровне каскада металлошаперонов, доставляющих медь АТФазе. Кроме того, спектр мутаций гена ATP7B имеет свои особенности проявления в различных популяциях [8, 13].
Наиболее распространена у европейских пациентов мутация H1069Q (доля мутации среди всех мутантных аллелей составляет 30–73% у пациентов с БВК в европеоидной популяции) [14]. В России замена H1069Q также является наиболее частой среди прочих мутантных аллелей в гене ATP7B (30–50%), при этом частота гетерозиготного носительства составляет 1 : 130 [15]. Другие часто встречаемые мутации в российской популяции – G126R и DelC3402 [8, 16, 17].
Мутация H1069Q представляет собой точечную замену C>A в положении 3027, в результате которой происходит замена гистидина на глутамин в высококонсервативном мотиве SEHPL белка ATP7B [18]. Это приводит к изменению пространственного расположения АТФ-связывающего сайта и нарушению фосфорилирования Р‑домена и, как следствие, уменьшению способности фермента связывать АТФ. Другая мутация в мотиве SEHPL, E1064K, ведет к полной потере связывания АТФ. У пациентов с этой мутацией БВК протекает особенно тяжело и сопровождается печеночной фульминантной недостаточностью [14, 19].
Помимо мутаций, инактивирующих ATP7B, существует относительно большое количество мутантных вариантов (G626A, D765N, M769V, I857T, A874V и др.), способных связывать и гидролизовать АТФ и обладающих частичной активностью транспорта меди. Этим объясняется фенотипическая гетерогенность и существование легких форм заболевания [10, 20]. Такие мутации могут приводить к более позднему началу заболевания и более легкому течению болезни. Следовательно, для лучшего понимания корреляции генотип–фенотип БВК, и более точного прогноза течения болезни у конкретного пациента, может потребоваться детальная характеристика каждого мутантного варианта ATP7B. Хотя мутации, расположенные в одном и том же домене, могут иметь сходные свойства, локализация только в определенном домене не является предиктором свойств мутации и степени тяжести болезни [10].
Вариабельность клинических проявлений БВК зависит, помимо прочего, от пола пациента [14]. Механизмом формирования этих различий считают влияние половых гормонов и метаболических процессов, особенно связанных с метаболизмом железа [21]. По результатам исследований 627 пациентов с болезнью Вильсона–Коновалова было выявлено, что неврологическая форма более присуща мужчинам, чем женщинам. При церебральной форме болезнь Вильсона–Коновалова у мужчин проявлялась в среднем на 2 года раньше, чем у женщин. А абдоминальная форма болезни Вильсона–Коновалова, как и различные гепатопатологии, чаще наблюдались у женщин [22]. Как полагают авторы исследования, мозг мужчин более чувствителен к накоплению меди, что может быть обусловлено разным уровнем половых гормонов. Одним из механизмов возникновения такого гендерного неравенства называют защитное действие эстрогенов [14]. Возможно, что у женщин с БВК эстрогены могут также защищать нейроны от накопления меди и окислительного стресса, который является следствием этого накопления, и таким образом ведут к уменьшению частоты проявления неврологических симптомов. Поэтому неврологические симптомы у мужчин проявляются раньше, чем у женщин, ведь у мужчин нет протективного действия эстрогенов [22]. Фактически влияние различий, связанных с полом, приводит к своеобразному модифицирующему эффекту на тяжесть и течение клинической картины БВК.
Влияние на клиническую картину болезни Вильсона–Коновалова, помимо мутаций в гене ATP7B, оказывают и определенные гены-модификаторы, являющиеся причиной формирования разных фенотипов у пациентов с идентичными генотипами [14, 23]. Гены-модификаторы – это группа генов, способных как утяжелять, так и облегчать фенотипические проявления генов, являющихся причиной заболевания. Генами-модификаторами при болезни Вильсона–Коновалова на сегодняшний день считаются ATP7A и APOE, в то же время существует ряд генов, функциональные характеристики белковых продуктов которых также указывают на возможность их включения в этот ряд (HFE, ATOX1, COMMD1, XIAP, PRNP, MTHFR, ESD, INO80).
ATP7A
Клетки человека экспрессируют две гомологичные Cu-АТФазы: ATP7A и ATP7B. Эти транспортеры используют энергию гидролиза АТФ для транспортировки меди из цитозоля через клеточные мембраны, тем самым снижая концентрацию цитозольной меди. Хотя и ATP7A, и ATB7B отвечают за экспорт меди, происходит он в разных типах клеток. При мутации в гене ATP7A клинические признаки являются последствиями нарушений медьзависимых ферментов. Дефицит меди вызывается дефектным ATP7A-опосредованным транспортом меди из кишечника в кровоток. Ген ATP7A экспрессируется почти во всех типах клеток и тканей, но уровни его экспрессии различаются в клетках и тканях и зависят от возраста [24], в то время как ген ATP7B преимущественно экспрессируется в печени взрослого человека [25]. Поэтому органом-мишенью при болезни Вильсона–Коновалова является печень – медь накапливается в печени в результате нарушенного ATP7B-обусловленного экспорта меди из гепатоцитов.
Мутации в гене ATP7A приводят к развитию болезни Менкеса – тяжелому Х-сцепленному заболеванию с дефицитом меди, которое обычно приводит к смерти больного в возрасте до 3 лет. Для этой болезни существует гендерное неравенство: преимущественно поражаются лица мужского пола, что объясняется гемизиготностью Х‑хромосомы у мужчин. В редких описанных случаях болезни Менкеса у женщин фенотипическое проявление болезни связывают с асимметричной инактивацией Х-хромосомы или же с наличием транслокационной формы заболевания [26, 27]. Так, описан единственный случай выявленной сбалансированной транслокации de novo 46,X,t(X;13)(q13.3;q14.3), при которой данная мутация затронула ATP7A (Xq13.3) и ATP7B (13q14.3) [28]. При этой мутации у женщины патологический фенотип соответствовал классической форме болезни Менкеса.
Поскольку ген ATP7A экспрессируется почти во всех клетках, патологический фенотип при поражении и гена ATP7A и гена ATP7B будет формироваться как при болезни Менкеса, и заболевание будет протекать значительно тяжелее, чем при классической болезни Вильсона–Коновалова. В литературе не описано случаев живорожденных детей с одновременной мутацией в генах ATP7A и ATP7B; вероятнее всего, такая патология несовместима с жизнью уже на стадии внутриутробного развития. К тому же недавние исследования показали, что ATP7A и ATP7B имеют различные и непересекающиеся функции в нейронах, экспрессирующих дофамин-β-гидроксилазу [29]. Соответственно ATP7A не обладает защитным или компенсаторным действием на метаболизм меди при БВК, а если на фоне мутации в гене ATP7B существуют мутации в гене ATP7A – это только усугубляет течение БВК.
HFE
Описаны редкие случаи сочетания болезни Вильсона–Коновалова с другими наследственными патологиями, в частности с наследственным гемохроматозом (НГХ). У пациентов с БВК были обнаружены, помимо мутации в гене АТР7В, также мутации в гене HFE, являющиеся причиной возникновения НГХ. Ген HFE (Homeostatic iron regulator) расположен в коротком плече шестой хромосомы и состоит из семи экзонов. Ген кодирует мембранный белок, подобный белкам главного комплекса гистосовместимости класса I и ассоциированный с бета2-микроглобулином (бета2М). Функция белка связана с регуляцией абсорбции железа путем взаимодействия трансферрина с рецептором [30]. Хотя вероятность возникновения мутаций одновременно в генах ATP7B и HFE невысока, описанные в литературе клинические случаи демонстрируют более раннее клиническое проявление заболевания Вильсона–Коновалова, чем при наличии только мутации в гене ATP7B [31–33]. В результате мутации происходит избыточное накопление железа в виде гемосидерина в клетках паренхиматозных органов, суставах и сердце, вследствие нарушения процессов всасывания железа в тонком кишечнике. Железо в свободной форме, как и медь, обладает токсическими свойствами, избыточное накопление этих металлов в организме приводит к образованию свободных радикалов и возникновению окислительного стресса, что в итоге приводит к разрушению клеток и нарушению функций жизненно важных органов. Кроме того, пути метаболизма меди и железа тесно пересекаются как в норме, так и при патологических состояниях [34]. Таким образом, сочетание двух связанных с нарушениями обмена металлов наследственных патологий приводит к более тяжелому поражению органов и течению заболевания в целом; следовательно, наличие мутации в гене HFE может рассматриваться как модифицирующий фактор болезни Вильсона–Коновалова.
ATOX1
Перенос меди из акцепторных белков обеспечивается уникальным классом белков, называемых медными шаперонами (такие как ATOX1 – Antioxidant 1 copper chaperone) [35], которые впервые были идентифицированы в дрожжах Saccharomyces cerevisiae. Ген ATOX1 (HAH1, ATX1) расположен на длинном плече хромосомы 5 (5q33.1), кодирует цитозольный белок массой 8 кДа и содержит четыре экзона. ATOX1 содержит одну N-концевую копию консервативного связывающего медь MXCXXC‑мотива, расположенного между первым бета-листом и альфа-спиралью, доставляет этот металл в транспортирующую медь АТФазу типа P-CCC2 в просвет транс-отдела сети Гольджи для последующей транспортировки по секреторному пути и включения в гомолог церулоплазмина – FET3. С помощью того же мотива ATOX1, помимо меди, связывает Hg(II), Cd(II), Ag(I) и цисплатин, но его физиологическая роль, если таковая имеется, пока не известна [36].
В клетках млекопитающих было показано, что белок ATOX1 доставляет медь к транспортирующим медь АТФазам – белкам болезни Менкеса и болезни Вильсона–Коновалова (ATP7A и ATP7B соответственно). Затем эти белки используют энергию гидролиза АТФ для того, чтобы либо перенести металл в просвет транс-отдела сети Гольджи для включения в медьзависимые ферменты, либо для вывода избытка меди из клетки. Выключение гена Atox1 у мышей приводит к внутриклеточному накоплению меди и снижению активности секретируемых медьзависимых ферментов, таких как тирозиназа, что подтверждает предполагаемую роль ATOX1 в качестве донора металла для транспортирующих медь АТФаз [37].
Существуют экспериментальные данные, показывающие наличие функциональной связи человеческого шаперона ATOX1 и транспортирующих медь АТФаз. Было показано, что некоторые мутации, обнаруженные у пациентов с БВК, нарушают способность ATP7B взаимодействовать с ATOX1, это позволяет предположить, что данное взаимодействие необходимо для нормального гомеостаза меди [38].
В недавнем исследовании 50 индийских пациентов с БВК N. Kumari с соавт. [40] выявили четыре новые мутации в гене ATOX1 (причем одна из них – c.40G>A, p.(G14S)), которые были идентифицированы в гетерозиготном состоянии у двух пациентов. Интересно, что при сравнении двух пациентов с аналогичными мутациями в гене ATP7B, но различающихся по статусу носительства мутации p.(G14S) в гене ATOX1, были выявлены существенные клинические различия: показано, что замена p.(G14S) ассоциирована с ранним возрастом начала заболевания, сниженным уровнем церулоплазмина в сыворотке и изменениями в печени и мозге у пациента с БВК в отличие от другого пациента, не имеющего мутаций в гене ATOX1 [40]. В то же время исследование данных секвенирования гена ATOX1 не выявило существенной связи или влияния на клиническое течение БВК [39].
Возможно, дальнейшие экспериментальные исследования на расширенных выборках, а также в различных популяциях дадут более четкую картину модификационной роли гена ATOX1 в патогенезе БВК.
COMMD1/XIAP
Одним из генов-кандидатов, которые могут играть роль гена-модификатора, является регулятор гомеостаза меди COMMD1 (copper metabolism domain containing 1, ранее известный как MURR1). Ген COMMD1 расположен на 2-й хромосоме (2p13), содержит восемь экзонов и кодирует растворимый белок массой 21 кДа, являющийся цитозольным белком и связывающий липиды с другими белками. COMMD1 – недавно идентифицированный фактор, который в основном участвует в двух процессах: регуляции фактора транскрипции NF-kB и контроле метаболизма меди. Впервые ген был упомянут в исследовании собак породы бедлингтон-терьеров, у которых отмечался токсикоз с симптомами высокого накопления меди в печени, что приводит к гепатиту и циррозу и возможной смерти, дальнейшие исследования выявили основную причину заболевания – делецию 2-го экзона гена Commd1, которая приводит к деградации белка [41, 42]. COMMD1 взаимодействует с N-терминальным доменом ATP7B, он может образовывать олигомерные гетерокомплексы с другими клеточными белками в дополнение к белку ATP7B [43].
Ген XIAP кодирует белок XIAP (X-linked inhibitor of apoptosis protein), относящийся к семейству белков – ингибиторов апоптоза (IAP) и обладающий выраженными антиапоптотическими свойствами [44–46]. Регуляция апоптоза осуществляется путем ингибирования специфических каспаз – цистеиновых протеаз [47]. Было показано, что XIAP, помимо основных своих функций, вовлечен в процесс регуляции гомеостаза меди через отрицательную регуляцию уровня белка COMMD1, по не связанному с апоптозом пути. Белок XIAP работает как регулятор уровня COMMD1, путем образования на нем полиубиквитиновых цепей, способствующих деградации белка COMMD1. Было показано, что повышение уровня экспрессии XIAP приводило к накоплению меди в клеточных моделях, а дефицит XIAP – к снижению содержания меди в тканях печени [48].
K.H. Weiss с соавт. [49] было проведено исследование групп пациентов с БВК и хотя бы одной подтвержденной мутацией в гене ATP7B: с пониженным и нормальным уровнем церулоплазмина, ни в одной из групп мутации в кодирующей последовательности гена COMMD1 обнаружено не было. Мутации в других частях гена COMMD1 не были проанализированы, но могли повлиять на экспрессию гена и таким образом повлиять на фенотип. По мнению авторов, выявление того, что ген COMMD1 не изменяется у пациентов с болезнью Вильсона–Коновалова с нормальным уровнем церулоплазмина в крови, необходимо оценить в более широком клиническом исследовании [49].
Таким образом, несмотря на немаловажную роль в регуляции метаболизма меди в организме, доказательств прямого влияния генов COMMD1 и XIAP на клиническое течение БВК получено не было.
АPOЕ
Один из известных генов-модификаторов – ген APOЕ, расположенный на хромосоме 19. Аполипопротеин Е (АпоЕ), кодируемый данным геном, вовлечен в метаболизм липидов в организме. Он является лигандом для нескольких типов рецепторов, в том числе для рецептора липопротеинов низкой плотности, который необходим для нормального катаболизма богатых триглицеридами липопротеинов. К основным функциям AпоE относится транспорт липидов, жирорастворимых липидов и холестерола через лимфатическую систему в кровь, что обеспечивает метаболизм холестерина. В крови AпоE регулирует всасывание остатков хиломикронов и остатков ЛПОНП печенью. В ЦНС он участвует в транспорте холестерина и других липидов к нейронам, обеспечивает их поглощение клетками. AпоE осуществляет доставку холестерина к месту миелинизации, что обусловливает его необходимость для поддержания миелиновой и нейрональной мембран как в центральной, так и в периферической нервной системе [50]. Существуют три основные изоформы белка – ε2, ε3 и ε4, встречающиеся в популяциях с частотами 6.4, 78.3 и 14.5% соответственно [51]. Структурные отличия в изоформах АпоЕ играют важную роль в развитии сердечно-сосудистых, нейродегенеративных и инфекционных заболеваний [52, 53]. Основной источник синтеза этого белка – печень (до 75%), вторым наиболее распространенным местом синтеза является мозг [54]. Макрофаги и другие типы клеток также синтезируют AпоE [52–54]. Ген APOE человека локализуется в хромосоме 19 и состоит из четырех экзонов, трех интронов, 3597 пар нуклеотидов, характеризуется высоким уровнем полиморфизма. Было показано, что данный ген оказывает влияние на клиническое проявление болезни Вильсона–Коновалова, в частности у пациентов с генотипом ε3/ε3 была отмечена манифестация болезни в более позднем возрасте [14, 55, 56].
В то же время другой группой исследователей [14, 22] была обнаружена связь аллельного варианта ɛ4 гена APOE с более ранним началом болезни Вильсона–Коновалова у женщин. Наиболее выражено влияние данного аллеля у женщин, являющихся носительницами мутации p.H1069G в гене ATP7B. Согласно полученным авторами данным, у этих пациенток болезнь Вильсона–Коновалова проявлялась на 6 лет раньше, чем у женщин с генотипом ε3/ε3 [14, 22].
V. Medici и K.H. Weiss [18] связывают аллель ε4 гена APOE с повышенной уязвимостью головного мозга в процессе развития болезни, тогда как генотип APOE ε3/3 обеспечивает умеренный нейропротективныйэффект.
PRNP
Ген PRNP находится на коротком плече 20-й хромосомы в локусе 20p13 и содержит два экзона. Ген кодирует прионный белок (PrPC) молекулярной массой 27–30 кДа. PrPC при помощи гликозилфосфатидилинозитола закреплен на внешней стороне клеточной мембраны и синтезируется в основном в клетках нервной системы и лимфоретикулярной ткани [14]. Его функция до конца не изучена, однако известно об участии PrPC в формировании миелиновой оболочки нервных волокон и процессах связывания низкоаффинных ионов меди, особенно в центральной нервной системе, где экспрессия белка наиболее высока [18, 57–59]. Было установлено, что взаимодействие меди с прионным белком может иметь нейропротективный эффект [60]. В другом исследовании была показана ассоциация замены гена PRNP в кодоне 129 (M129V) с началом и последующим развитием болезни Вильсона–Коновалова [61]. Согласно полученным исследователями данным, распространенность генотипа M129V у пациентов с БВК была сходной с контрольной популяционной выборкой. Также было показано влияние полиморфных вариантов PRNP на клиническую картину БВК: у пациентов с вариантом PRNP 129M, приводящим к гомозиготному метионину, возраст начала болезни был примерно на 5 лет выше, с возникновением неврологических симптомов в среднем через 7 лет после начала болезни, по сравнению с носителями гетерозиготного варианта.
В то же время оказывать влияние на фенотип при болезни Вильсона–Коновалова могут и прионные болезни (OMIM #123400, #137440, #600072). Прионные болезни возникают, как правило, спорадически, либо в очень редких случаях в результате инфекции, но также могут возникать (в 5–15% случаев) в связи с фенотипически доминантными мутациями в гене PRNP. Прионные болезни, так же как и болезнь Вильсона–Коновалова, приводят к прогрессирующей необратимой нейродегенерации с когнитивными и психиатрическими проявлениями. N. Forbes с соавт. [62] был описан клинический случай пациента с быстро прогрессирующими неврологическими симптомами БВК. У данного пациента были обнаружены несинонимичные варианты кодирующей последовательности в генах ATP7B и PRNP. Авторы считают, что именно синергетическое взаимодействие между наблюдаемыми вариантами ATP7B и PRNP, опосредованное эффектами кодируемых ими белков на метаболизм меди, было причиной ухудшения течения болезни пациента [62].
MTHFR
Еще одним геном, для которого показано влияние на возраст манифестации БВК, является ген MTHFR (хромосомная локализация 1р36.3), который состоит из 20 200 пар нуклеотидов и содержит 11 экзонов. Этот ген кодирует метилентетрагидрофолатредуктазу (MTHFR) – внутриклеточный фермент, играющий роль в метаболизме гомоцистеина и фолата. Фермент катализирует восстановление 5,10-метилентетрагидрофолата в 5-метилтетрагидрофолат. Последний является активной формой фолиевой кислоты, необходимой для образования метионина из гомоцистеина и далее – S-аденозилметионина, играющего ключевую роль в процессе метилирования ДНК.
Наиболее значимые с точки зрения патогенеза болезни Вильсона–Коновалова полиморфные варианты гена MTHFR – С677Т и A1298C. Данные замены приводят к снижению активности метилентетрагидрофолатредуктазы и, как следствие, развитию гипергомоцистеинемии. Вариант С677Т гена MTHFR представляет собой замену цитозина на тимидин в позиции 677, что, в свою очередь, приводит к замене аланина на валин в апобелке этого фермента. А1298С – полиморфизм гена MTHFR с заменой аденина на цитозин в позиции 1298, не сопровождающийся повышением уровня гомоцистеина в крови, однако сочетание гетерозиготного носительства аллелей 677Т и 1298С сопровождается повышением уровня гомоцистеина в плазме и приводит к снижению активности фермента MTHFR. Установлено, что повышение уровня гомоцистеина влияет на гомеостаз меди в организме и внутриклеточную токсичность ионов меди [63, 64]. Кроме того, значительное повышение концентрации гомоцистеина в клетках приводит к сильному окислительному стрессу и клеточной гибели [65].
G. Gromadzka и соавт. [66] предположили, что гепатотоксические эффекты при болезни Вильсона–Коновалова могут быть более выраженными, чем при патологиях печени другой этиологии, так как гомоцистеин и медь проявляют сильные синергические цитотоксические эффекты. Кроме того, этой же группой исследователей была установлена связь полиморфных вариантов гена MTHFR с возрастом манифестации БВК. У носителей гомозиготного варианта 1298C начало заболевания фиксировалось примерно на 6 лет раньше, чем у пациентов с генотипом 677CC/1298AA. На основании полученных данных авторы предполагают, что гомоцистеин может также быть вовлечен в нейродегенеративные процессы при БВК [66].
ESD и INO80
Помимо вышеописанных генов, влияние которых на фенотип БВК достаточно изучено и однозначно, существуют гены, о которых известно только то, что они модифицируют течение БВК, но информация о механизмах их влияния либо отсутствует, либо не является исчерпывающей. К таким малоизученным, но, несомненно, важным для патогенеза БВК генам относятся ESD и INO80.
Недавние исследования показали, что при редких аллельных вариантах ESD значительно чаще отмечалось появление у пациентов неврологической симптоматики, в то время как эффекты вариантов в гене INO80 были прямо противоположными – неврологические нарушения БВК возникали гораздо реже [67].
Ген ESD расположен в длинном плече хромосомы 13 и состоит из 11 экзонов. Ген кодирует сериновую гидролазу, которая принадлежит семейству эстеразы D. Кодируемый фермент активен по отношению к многочисленным субстратам, включая O-ацетилированные сиаловые кислоты, и может участвовать в рециркуляции сиаловых кислот. Ген используется в качестве генетического маркера для ретинобластомы и БВК [68]. Влияние гена ESD на фенотипические проявления БВК исследователи объясняют его близким хромосомным расположением к ATP7B и возможным сцеплением между ними, однако точных доказательств этой гипотезы предложено не было [67, 69].
Ген INO80 расположен в длинном плече хромосомы 15, состоит из 37 экзонов. Кодирует INO80 – каталитическую АТФазную субъединицу комплекса ремоделирования хроматина, которая принимает участие в процессах транскрипции, репарации и репликации ДНК. О конкретных механизмах участия продуктов данного гена на патогенез болезни Вильсона–Коновалова известно мало, но предполагается, что он оказывает влияние на поддержание стабильности ДНК путем увеличения подвижности хроматина, тем самым оказывая нейропротективное действие (в том числе при окислительном повреждении клеток нервной системы, сопровождающем болезнь Вильсона–Коновалова) [70, 71].
Таким образом, вариабельность клинических фенотипов при болезни Вильсона–Коновалова может быть обусловлена различными мутациями не только в гене ATP7B, но и в тех генах, которые участвуют в гомеостазе меди, а также различиями, связанными с полом (табл. 1).
Таблица 1.
Участие генов в патогенезе болезни Вильсона–Коновалова и их эффект
| № | Ген | Локализация на хромосомах | Продукт гена | Влияние на течение БВК, источник |
|---|---|---|---|---|
| 1 | ATP7A | Хq12-q13 | Белок ATP7A | ATP7A и ATP7B имеют различные и непересекающиеся функции в нейронах, экспрессирующих дофамин-β-гидроксилазу [27] |
| 2 | ATOX1 | 5q33.1 | Белок ATOX1 | p.(G14S) была ассоциирована с ранним возрастом начала заболевания, сниженным уровнем церулоплазмина в сыворотке, изменениями в печени и мозге [38] |
| 3 | COMMD1 | 2p13 | Регулятор гомеостаза меди COMMD1 | Гетерозиготное состояние GAT/GAC в кодоне Asn164 связано с более ранним началом заболевания [47] |
| 4 | XIAP | Xq25 | X-связанный ингибитор апоптоза | Повышение уровня экспрессии XIAP приводит к накоплению меди в клеточных моделях, а дефицит – к снижению содержания меди в тканях печени [46] |
| 5 | HFE | 6p22.2 | Белок HFE | Усиливает окислительный стресс и повреждение клеток [29] |
| 6 | APOE | 19q13.32 | Белок АпоЕ | Нейропротективный эффект генотипа ɛ3/ɛ3 [57, 58], усиливающий повреждение нейронов генотип ɛ4 [16] |
| 7 | PRNP | 20p13 | Прионный белок PrPC | Нейропротективный эффект варианта 129 M/M [62] |
| 8 | MTHFR | 1р36.3 | Метилентетрагидрофолатредуктаза | Более выраженные гепатотоксические и нейродегенеративные процессы [68] |
| 9 | ESD | 13q14.2 | Сериновая гидролаза из семейства эстеразы D | Более выраженные нейродегенеративные процессы [69] |
| 10 | INO80 | 15q15.1 | Белок INO80 | Нейропротективный эффект [71] |
К фенотипически более легкому течению болезни Вильсона–Коновалова (преимущественно за счет нейропротективного эффекта) предрасполагают полиморфные варианты генов APOE (ε3/ε3), PRNP, INO80. В то же время полиморфизм генов APOE (ε4), COMMD1, ATOX1, HFE, MTHFR и ESD, напротив, может усугубить течение и симптоматику болезни Вильсона–Коновалова.
Дальнейшее изучение причин клинического полиморфизма и различий начала манифестации болезни Вильсона–Коновалова важно для практической медицины, поскольку может помочь врачу поставить верный диагноз и обосновать выбор молекулярно-генетического метода для подтверждения этиологии заболевания и соответственно скорректировать лечение в рамках персонализированной медицины.
Исследование выполнено в рамках государственного задания на фундаментальные научные исследования НИИ медицинской генетики Федерального государственного бюджетного научного учреждения “Томский национальный исследовательский медицинский центр Российской академии наук” по теме “Персонализированное геномное профилирование при орфанных заболеваниях человека” (№ АААА-А19-119090990020-0).
Настоящая статья не содержит каких-либо исследований с использованием в качестве объекта животных.
Настоящая статья не содержит каких-либо исследований с участием в качестве объекта людей.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Асанов А.Ю., Соколов А.А., Волгина С.Я. и др. Федеральные клинические рекомендации по диагностике и лечению болезни Вильсона–Коновалова. М., 2015. 71 с.
Корягина О.Ю., Назаренко Л.П., Филимонова М.Н. Гепатолентикулярная дегенерация // Сиб. бюл. 2005. C. 61.
Poujois A., Woimant F. Wilson’s disease: A 2017 update // Clin. Res. Hepatol. Gastroenterol. 2018. V. 42. № 6. P. 512–520. https://doi.org/10.1016/j.clinre.2018.03.007
Coffey A.J., Durkie M., Hague S. et al. A genetic study of Wilson’s disease in the United Kingdom // Brain. 2013. V. 136. № 5. P. 1476–1487. https://doi.org/10.1093/brain/awt035
Баязутдинова Г.М., Щагина О.А., Карунас А.С. и др. Спектр мутаций в гене ATP7B у российских больных с болезнью Вильсона–Коновалова // Генетика. 2019. Т. 55. № 12. С. 1433–1441. https://doi.org/10.1134/s0016675819120026
Ala A., Walker A.P., Ashkan K. et al. Wilson’s disease // The Lancet. 2007. V. 369. № 9559. P. 397–408. https://doi.org/10.1016/S0140-6736(07)60196-2
De Bie P., Muller P., Wijmenga C., Klomp L.W.J. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes // J. Med. Genet. 2007. V. 44. № 11. P. 673–688. https://doi.org/10.1136/jmg.2007.052746
Тулузановская И.Г., Балашова М.С., Жученко Н.А. и др. Мутации в гене HFE, ответственном за наследственный гемохроматоз, у пациентов с болезнью Вильсона–Коновалова // Эксперим. и клинич. гастроэнтерология. 2018. № 11(159). С. 33.
Maxwell K.L., Kowdley K.V. Metals and the liver // Curr. Opin. Gastroenterol. 2012. V. 28. № 3. P. 217–222. https://doi.org/10.1097/MOG.0b013e3283521d82
Huster D., Kühne A., Bhattacharjee A. et al. Diverse functional properties of Wilson disease ATP7B variants // Gastroenterology. 2012. V. 142. № 4. P. 947–956. e5. https://doi.org/10.1053/j.gastro.2011.12.048
Okada T., Shiono Y., Kaneko Y. et al. High prevalence of fulminant hepatic failure among patients with mutant alleles for truncation of ATP7B in Wilson’s disease // Scand. J. Gastroenterol. 2010. V. 45. № 10. P. 1232–1237. https://doi.org/10.3109/00365521.2010.492527
Gromadzka G., Schmidt H.J., Genschel J. et al. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease // Clin. Genet. 2005. V. 68. № 6. P. 524–532. https://doi.org/10.1111/j.1399-0004.2005.00528
Perri R.E., Hahn S.H., Ferber M.J. et al. Wilson disease – keeping the bar for diagnosis raised // Hepatology. 2005. V. 42. № 4. P. 974. https://doi.org/10.1002/hep.20893
Баязутдинова Г.М., Щагина О.А., Поляков А.В. Молекулярный патогенез болезни Вильсона–Коновалова // Медицинская генетика. 2017. Т. 16. № 7. С. 18–24.
Баязутдинова Г.М., Щагина О.А., Поляков А.В. Мутация с.3207C>A гена ATP7B – наиболее частая причина гепатолентикулярной дегенерации в России: частота и причина распространения // Мед. генетика. 2018. T. 17(4). C. 25–30.
Ivanova-Smolenskaya I.A., Ovchinnikov I.V., Karabanov A.V. et al. The His1069Gln mutation in the ATP7B gene in Russian patients with Wilson disease // J. Med. Genet. 1999. V. 36. № 2. P. 174. https://doi.org/10.1136/jmg.36.2.174
Matveeva T., Zaklyazminskaya E., Polyakov A. The molecular-genetic analysis of ATP7B gene at the Russian patients with Wilson disease // Eur. J. Hum. Genet. 2008. V. 16. P. 54.
Medici V., Weiss K.H. Genetic and environmental modifiers of Wilson disease // Handb. Clin. Neurol. 2017. V. 142. P. 35–41.
Firneisz G., Szonyi L., Ferenci P. et al. The other mutation is found: follow-up of an exceptional family with Wilson disease // The Am. J. Gastroenterol. 2004. V. 99. № 12. P. 2504. https://doi.org/10.1111/j.1572-0241.2004.41389_8.x
Lutsenko S. Modifying factors and phenotypic diversity in Wilson’s disease // Ann. N.Y. Acad. Sci. 2014. V. 1315. P. 56. https://doi.org/10.1111/nyas.12420
Shulman L.M. Gender differences in Parkinson’s disease // Gender Med. 2007. V. 4. № 1. P. 8–18. https://doi.org/10.1016/S1550-8579(07)80003-9
Litwin T., Gromadzka G., Członkowska A. Gender differences in Wilson’s disease // J. Neurol. Sci. 2012. V. 312. № 1–2. P. 31–35. https://doi.org/10.1016/j.jns.2011.08.028
Chen C., Shen B., Xiao J.J. et al. Currently clinical views on genetics of Wilson’s disease // Chin. Med. J. 2015. V. 128. № 13. P. 1826. https://doi.org/10.4103/0366-6999.159361
Lenartowicz M., Wieczerzak K., Krzeptowski W. et al. Developmental changes in the expression of the Atp7a gene in the liver of mice during the postnatal period // J. Exp. Zool. A. Ecol. Genet. Physiol. 2010. V. 313. № 4. P. 209–217. https://doi.org/10.1002/jez.586
Ogórek M., Lenartowicz M., Starzyński R. et al. Atp7a and Atp7b regulate copper homeostasis in developing male germ cells in mice //Metallomics. 2017. V. 9. № 9. P. 1288–1303. https://doi.org/10.1039/c7mt00134g
Lenartowicz M., Moos T., Ogórek M. et al. Metal-dependent regulation of ATP7A and ATP7B in fibroblast cultures // Front. Mol. Neurosci. 2016. V. 9. P. 68. https://doi.org/10.3389/fnmol.2016.00068
Desai V., Donsante A., Swoboda K.J. et al. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease // Clin. Genet. 2011. V. 79. № 2. P. 176–182. https://doi.org/10.1111/j.1399-0004.2010.01451.x
Abusaad I., Mohammed S.N., Ogilvie C.M. et al. Clinical expression of Menkes disease in a girl with X; 13 translocation // Am. J. Med. Genet. 1999. V. 87. № 4. P. 354–359.
Schmidt K., Ralle M., Schaffer T. et al. ATP7A and ATP7B copper transporters have distinct functions in the regulation of neuronal dopamine-β-hydroxylase // J. Biol. Chem. 2018. V. 293. № 52. P. 20085–20098. https://doi.org/10.1074/jbc.RA118.004889
Pruitt K.D., Tatusova T., Klimke W., Maglott D.R. NCBI reference sequences: current status, policy and new initiatives // Nucl. Acids Res. 2009. V. 37. Suppl. 1. P. D32–D36. https://doi.org/10.1093/nar/gkn721
Ala A., Schilsky M. Genetic modifiers of liver injury in hereditary liver disease // Semin. Liver Dis. 2011. V. 31. № 02. P. 208–214. https://doi.org/10.1055/s-0031-1276648
Abuzetun J.Y., Hazin R., Suker M., Porter J. A rare case of hemochromatosis and Wilson’s disease coexisting in the same patient // J. Natl Med. Assoc. 2008. V. 100. № 1. P. 112. https://doi.org/10.1016/s0027-9684(15)31185-8
Dib N., Valsesia E., Malinge M.C. et al. Late onset of Wilson’s disease in a family with genetic haemochromatosis // Eur. J. Gastroenterol. Hepatol. 2006. V. 18. № 1. P. 43–47. https://doi.org/10.1097/00042737-200601000-00008
Багаева М.Э., Каганов Б.С., Готье С.В. и др. Клиническая картина и течение болезни Вильсона у детей // Вопр. соврем. педиатрии. 2004. Т. 3. № 5. С. 13–18.
Culotta V.C., Klomp L.W.J., Strain J. et al. The copper chaperone for superoxide dismutase // J. Biol. Chem. 1997. V. 272. № 38. P. 23469–23472. https://doi.org/10.1074/jbc.272.38.23469
Maret W., Wedd A. Binding, transport and storage of metal ions in biological cells // Royal Society Chem. 2014. V. 2. https://doi.org/10.1039/9781849739979
Hamza I., Faisst A., Prohaska J. et al. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis // PNAS. 2001. V. 98. № 12. P. 6848–6852. https://doi.org/10.1073/pnas.111058498
Hamza I., Schaefer M., Klomp L.W.J. et al. Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis // PNAS USA. 1999. V. 96. № 23. P. 13363–13368. https://doi.org/10.1073/pnas.96.23.13363
Simon I., Schaefer M., Reichert J., Stremmel W. Analysis of the human Atox 1 homologue in Wilson patients // World J. Gastroenterol. 2008. V. 14. № 15. P. 2383. https://doi.org/10.3748/wjg.14.2383
Kumari N., Kumar A., Pal A. et al. In silico analysis of novel p.(Gly14Ser) variant of ATOX1 gene: plausible role in modulating ATOX1–ATP7B interaction // Mol. Biol. Rep. 2019. V. 46. № 3. P. 3307–3313. https://doi.org/10.1007/s11033-019-04791-x
Brewer G.J., Dick R.D., Schall W. et al. Use of zinc acetate to treat copper toxicosis in dogs // J. Am. Vet. Med. Assoc. 1992. V. 201. № 4. P. 564.
van de Sluis B., Rothuizen J., Pearson P.L. et al. Identification of a new copper metabolism gene by positional cloning in a purebred dog population // Hum. Mol. Genet. 2002. V. 11. № 2. P. 165–173. https://doi.org/10.1093/hmg/11.2.165
Tao T.Y., Liu F., Klomp L. et al. The copper toxicosis gene product Murr1 directly interacts with the Wilson disease protein // J. Biol. Chem. 2003. V. 278. № 43. P. 41593–41596. https://doi.org/10.1074/jbc.C300391200
Chai J., Shiozaki E., Srinivasula S.M. et al. Structural basis of caspase-7 inhibition by XIAP // Cell. 2001. V. 104. № 5. P. 769–780. https://doi.org/10.1016/S0092-8674(01)00272-0
Huang Y., Park Y.C., Rich R.L. et al. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain // Cell. 2001. V. 104. № 5. P. 781–790. https://doi.org/10.1016/s0092-8674(02)02075-5
Riedl S.J., Renatus M., Schwarzenbacher R. et al. Structural basis for the inhibition of caspase-3 by XIAP // Cell. 2001. V. 104. № 5. P. 791–800. https://doi.org/10.1016/S0092-8674(01)00274-4
Lewis J., Burstein E., Reffey S.B. et al. Uncoupling of the signaling and caspase-inhibitory properties of X-linked inhibitor of apoptosis // J. Biol. Chem. 2004. V. 279. № 10. P. 9023–9029. https://doi.org/10.1074/jbc.M312891200
Harlin H., Reffey S.B., Duckett C.S. et al. Characterization of XIAP-deficient mice // Mol. Cell. Biol. 2001. V. 21. № 10. P. 3604–3608. https://doi.org/10.1128/mcb.21.10.3604-3608.2001
Weiss K.H., Merle U., Schaefer M. et al. Copper toxicosis gene MURR1 is not changed in Wilson disease patients with normal blood ceruloplasmin levels // World J. Gastroenterol. 2006. V. 12. № 14. P. 2239. https://doi.org/10.3748/wjg.v12.i14.2239
Leduc V., Jasmin-Bélanger S., Poirier J. APOE and cholesterol homeostasis in Alzheimer’s disease // Trends Mol. Med. 2010. V. 16. № 10. P. 469–477. https://doi.org/10.1016/j.molmed.2010.07.008
Eisenberg D.T.A., Kuzawa C.W., Hayes M.G. Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history // Am. J. Phys. Anthropol. 2010. V. 143. № 1. P. 100–111. https://doi.org/10.1002/ajpa.21298
Mahley R.W., Rall Jr S.C. Apolipoprotein E: far more than a lipid transport protein // Annu. Rev. Genomics Hum. Genet. 2000. V. 1. № 1. P. 507–537. https://doi.org/10.1146/annurev.genom.1.1.507
Harris F.M., Brecht W.J., Xu Q. et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice // PNAS. 2003. V. 100. № 19. P. 10966–10971. https://doi.org/10.1073/pnas.1434398100
Elshourbagy N.A., Liao W.S., Mahley R.W., Taylor J.M. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets // PNAS USA. 1985. V. 82. № 1. P. 203–207. https://doi.org/10.1073/pnas.82.1.203
Członkowska A., Gromadzka G., Chabik G. Monozygotic female twins discordant for phenotype of Wilson’s disease // Mov. Disord. 2009. V. 24. № 7. P. 1066–1069. https://doi.org/10.1002/mds.22474
Wang X.P., Wang X.H., Bao Y.C., Zhou J.N. Apolipoprotein E genotypes in Chinese patients with Wilson’s disease // QJM. 2003. V. 96. № 7. P. 541–542. https://doi.org/10.1093/qjmed/hcg093
Ford M.J., Burton L.J., Morris R.J., Hall S.M. Selective expression of prion protein in peripheral tissues of the adult mouse // Neuroscience. 2002. V. 113. № 1. P. 177–192. https://doi.org/10.1016/S0306-4522(02)00155-0
Brown D.R., Qin K., Herms J.W. et al. The cellular prion protein binds copper in vivo // Nature. 1997. V. 390. № 6661. P. 684–687. https://doi.org/10.1038/37783
Bremer J., Baumann F., Tiberi C. et al. Axonal prion protein is required for peripheral myelin maintenance // Nat. Neurosci. 2010. V. 13. № 3. P. 310–318. https://doi.org/10.1038/nn.2483
Gasperini L., Meneghetti E., Pastore B. et al. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation // Antioxid. Redox Signal. 2015. V. 22. № 9. P. 772–784. https://doi.org/10.1089/ars.2014.6032
Merle U., Stremmel W., Geßner R. Influence of homozygosity for methionine at codon 129 of the human prion gene on the onset of neurological and hepatic symptoms in Wilson disease // Arch. Neurol. 2006. V. 63. № 7. P. 982–985. https://doi.org/10.1001/archneur.63.7.982
Forbes N., Goodwin S., Woodward K. et al. Evidence for synergistic effects of PRNP and ATP7B mutations in severe neuropsychiatric deterioration // BMC Med. Genet. 2014. V. 15. № 1. P. 1–6. https://doi.org/10.1186/1471-2350-15-22
Tamura T., Picciano M.F. Folate and human reproduction // Am. J. Clin. Nutr. 2006. V. 83. № 5. P. 993–1016. https://doi.org/10.1093/ajcn/83.5.993
Friso S., Girelli D., Trabetti E. et al. The MTHFR 1298A>C polymorphism and genomic DNA methylation in human lymphocytes // Cancer Epidemiol. Biomarkers Prev. 2005. V. 14. № 4. P. 938–943. https://doi.org/10.1158/1055-9965.EPI-04-0601
Мещерякова Т.И., Маркова С.И., Жилина С.С. и др. Изучение влияния полиморфизма C677T гена MTHFR на риск формирования несиндромальных орофациальных расщелин // Рос. вестник перинатологии и педиатрии. 2013. T. 58. № 3.
Gromadzka G., Rudnicka M., Chabik G. et al. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson’s disease // J. Hepatol. 2011. V. 55. № 4. P. 913–919. https://doi.org/10.1016/j.jhep.2011.01.030
Kluska A.,Kulecka M., Litwin T. et al. Whole-exome sequencing identifies novel pathogenic variants across the ATP7B gene and some modifiers of Wilson’s disease phenotype // Liver Int. 2019. V. 39. № 1. P. 177–186. https://doi.org/10.1111/liv.13967
Frydman M., Bonné-Tamir B., Farrer L.A. et al. Assignment of the gene for Wilson disease to chromosome 13: Linkage to the esterase D locus // PNAS USA. 1985. V. 82. № 6. P. 1819–1821. https://doi.org/10.1073/pnas.82.6.1819
Chuang L.M., Tai T.Y., Wang T.R. et al. Esterase D and retinoblastoma gene loci are tightly linked to Wilson’s disease in Chinese pedigrees from Taiwan // Hum. Genet. 1991. V. 87. № 4. P. 465–468. https://doi.org/10.1007/BF00197170
Poli J., Gasser S.M., Papamichos-Chronakis M. The INO80 remodeller in transcription, replication and repair // Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017. V. 372. № 1731. P. 20160290. https://doi.org/10.1098/rstb.2016.0290
Medici V., LaSalle J.M. Genetics and epigenetic factors of Wilson disease // Ann. Transl. Med. 2019. V. 7. Suppl. 2. https://doi.org/10.21037/atm.2019.01.67
Дополнительные материалы отсутствуют.