

Генетика, 2021, T. 57, № 8, стр. 949-954
Новая миссенс-мутация Gly238Ala в гене TBX5 и ее фенотипическая характеристика
Н. Н. Чакова 1, *, Т. В. Долматович 1, С. С. Ниязова 1, С. М. Комиссарова 2, Е. С. Ребеко 2, А. А. Савченко 2
1 Институт генетики и цитологии Национальной академии наук Беларуси
220072 Минск, Республика Беларусь
2 Республиканский научно-практический центр “Кардиология”
220036 Минск, Республика Беларусь
* E-mail: n.chakova@igc.by
Поступила в редакцию 18.09.2020
После доработки 23.11.2020
Принята к публикации 14.12.2020
Аннотация
Ген TBX5 кодирует Т-box содержащий транскрипционный фактор 5 (Tbx5), который регулирует процесс эмбриогенеза у позвоночных и беспозвоночных. Мутации в этом гене являются причиной развития редкого моногенного синдрома Холт–Орама (HOS), характеризующегося скелетными аномалиями верхних конечностей, врожденным пороком сердца и/или нарушениями проводящей системы миокарда. Методом высокопроизводительного секвенирования (NGS) выявлен новый нуклеотидный вариант c.713G>C (p.Gly238Ala) в гене TBX5 у пациентки с мягким фенотипом синдрома Холта–Орама (деформация грудного отдела позвоночника (сколиоз), дисплазия лопатки, недостаточность митрального и трикуспидального клапанов с регургитацией I–II степени и истончение межпредсердной перегородки) и угрожающими жизни тахиаритмиями, потребовавшими имплантации кардиовертера-дефибриллятора (ИКД). Мутация p.Gly238Ala локализована в “горячей точке” гена TBX5. Оценка ее патогенности методом анализа in silico показала, что нуклеотидная замена c.713G>C может приводить к изменению структуры и/или функции белка. Устойчивая желудочковая тахикардия/фибрилляция желудочков, которые не являются характерной чертой HOS, могли быть результатом двух дополнительных редких замен (MAF < 0.01%): p.Val3634Asp, rs66785829 в гене ANK2 и p.Arg1193Gln, rs41261344 в гене SCN5A. Мутации в этих генах влияют на функционирование натриевого ионного канала и являются причиной наследственных аритмий.
Ген TBX5, расположенный на хромосоме 12q24.21, кодирует Т-box-содержащий эволюционно консервативный транскрипционный фактор 5 (Tbx5), который регулирует широкий спектр процессов эмбриогенеза у позвоночных и беспозвоночных, включая спецификацию мезодермы и развитие сердечно-сосудистой системы и конечностей [1, 2]. У людей и позвоночных Tbx5 экспрессируется в эпикарде, миокарде всех четырех камер сердца, эндокарде левого желудочка [3] и атриовентрикулярном канале и играет ключевую роль как в структурной организации миокарда, так и в формировании его проводящей системы [4]. Tbx5 является членом семейства транскрипционных факторов, характеризующихся высококонсервативным ДНК-связывающим доменом T‑box [5], который состоит из 180 аминокислот. Показано, что Tbx5 посредством специфического связывания с ДНК активирует транскрипцию генов ANF, CX40 и SRF, которые могут по отдельности или совместно работать с белками NKX2-5, GATA4 и TBX20 [1].
Мутации в гене TBX5 являются причиной развития редкого моногенного синдрома Холт–Орама (HOS, MIM 142900), характеризующегося скелетными аномалиями верхних конечностей, наличием врожденного порока сердца (ВПС), чаще всего представленного дефектом межпредсердной перегородки, и/или нарушениями проводящей системы миокарда. Заболевание представляет собой плейотропное расстройство с полной пенетрантностью, но с различной экспрессией (выраженностью) даже в одной семье [6]. Около 85% пациентов имеют изменения сразу в обеих системах организма [7].
HOS является наиболее распространенным синдромом “сердце-рука”. Этот синдром был впервые описан М. Холт и С. Орамом в 1960 г. [8]. Распространенность HОS у новорожденных составляет приблизительно 0.7 на 100 000 рождений и не имеет гендерных различий [9]. В 1997 г. выявлены первые мутации в гене TBX5 у пациентов с HOS [10, 11]. К настоящему моменту идентифицировано более 90 мутаций [12], локализованных преимущественно в 3–7 экзонах, соответствующих области домена T-box. Практически 87% представлены точковыми изменениями, большинство из которых (37% – нонсенс-мутации, 26% – мутации, вызывающие сдвиг рамки считывания, 10% – мутации в сайтах сплайсинга) действуют по принципу гаплонедостаточности за счет синтеза нефункционального укороченного белка или запуска нонсенс-опосредованного распада мРНК и обычно сопровождаются тяжелыми пороками развития верхних конечностей и сердца [13]. На долю обширных внутригенных делеций и дупликаций в гене TBX5 приходится 8 и 4% соответственно. Описаны также сбалансированные транслокации с участием локуса TBX5 [13]. Миссенс-мутации по разным данным составляют 14–30%, обладают доминантно-негативным эффектом и являются причиной синтеза белков со сниженной ДНК-связывающей активностью. Некоторые исследователи показали, что аминокислотная замена на амино-конце Т-box приводит к очень значительным порокам развития сердца и небольшим аномалиям скелета, а изменение аминокислот на карбоксильном конце наоборот вызывает тяжелые аномалии конечностей и менее значимые сердечные аномалии [7]. Однако существует и противоположное мнение, что ни тип мутации в TBX5, ни место мутации в Т-box не являются предикторами выраженности пороков развития [14].
По результатам ряда исследований, мутации в гене TBX5 выявлялись у 36–70% пациентов с клиническими признаками синдрома HOS [13, 15, 16]. У 78% пациентов с мутациями фенотип заболевания строго соответствовал диагностическим критериям HOS, в 20% случаев имелась менее выраженная клиническая симптоматика [13]. Большинство мутаций возникает de novo [6]. На данный момент базы данных по мутациям в этом гене продолжают активно пополняться новыми вариантами [5, 7].
В настоящей статье мы также сообщаем о не встречавшейся ранее в мировой популяции мутации c.713G>C, приводящей к изменению аминокислоты p.Gly238Ala в области ДНК-связывающего домена T-box транскрипционного фактора Tbx5, выявленной у пациентки с мягким фенотипом HOS и наличием жизнеугрожающих аритмий (устойчивая желудочковая тахикардия/фибрилляции желудочков).
МАТЕРИАЛЫ И МЕТОДЫ
Пациентка А. с жизнеугрожающими желудочковыми аритмиями и синкопальными состояниями наблюдалась в РНПЦ “Кардиология” г. Минска, Беларусь. Клиническое обследование включало ЭКГ в 12 отведениях, ЭхоКГ, МРТ сердца с отсроченным контрастированием и суточное мониторирование ЭКГ (СМ ЭКГ). Получено информированное согласие пациентки на участие в научном исследовании.
Для определения генетической причины нарушения ритма пациентке было выполнено генетическое тестирование методом высокопроизводительного секвенирования (NGS) с использованием набора реагентов “TruSight™ Cardio Sequencing Panel” (Illumina), включающего кодирующие последовательности 174 генов, ассоциированных с наследственными сердечно-сосудистыми заболеваниями, на приборе MiSeq (Illumina). Обработка и аннотирование результатов секвенирования проводились с помощью специального программного обеспечения ANNOVAR rev. 527 [17], позволяющего оценить патогенность выявленного генетического варианта на основе баз данных (dbSNP, 1000genomes, GWAS, HGMD и др.), и предсказательных модулей (PolyPhen-2 [18], SIFT [19], FATHMM [20] и Mutationtester [21]).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Поиск мутаций проводился у 54-летней женщины, которая в 42 года внезапно потеряла сознание на фоне участившихся сердцебиений, что потребовало проведение реанимационных мероприятий и имплантации кардиовертера-дефибриллятора (ИКД). На ЭКГ был зафиксирован эпизод желудочковой тахикардии/фибрилляции желудочков (ЖТ/ФЖ). Сердцебиения и синкопальные состояния отмечались с 6-летнего возраста. По данным ЭхоКГ камеры сердца не расширены, нарушений сократительной способности миокарда левого желудочка (ЛЖ) не выявлено. Фракция выброса ЛЖ составила 63%. Зарегистрирована митральная и трикуспидальная регургитация I–II степени и истончение межпредсердной перегородки в средней трети. ВПС не обнаружено.
По данным суточного мониторирования ЭКГ отмечалась частая желудочковая экстрасистолия (1860 за сутки) с неустойчивыми пробежками полиморфной желудочковой тахикардии (ЖТ). На фоне антиаритмической терапии (бисопролол 5 мг/сутки) эпизоды неустойчивой, устойчивой ЖТ и фибрилляции желудочка (ФЖ) сохранялись. По данным монитора ИКД выявлен эпизод фибрилляции желудочков, успешно купированный разрядом в 35 Дж.
В результате генетического тестирования у пациентки обнаружена новая, ранее не описанная миссенс-мутация c.713G>C в экзоне 7 гена TBX5, которая приводит к замене глицина на аланин (p.Gly238Ala). Мутация расположена в карбоксильной области ДНК-связывающего домена T-box. Остаток глицина высококонсервативен у разных видов животных и сохраняется в гомологичном белке Tbx4 человека (рис. 1). У всех других членов семейства T-box-содержащих белков, таких как Tbx1, Tbx2, Tbx3 и Tbx20, в этом положении находится аспарагин (D), что также указывает на достаточную консервативность данного региона.
Рис. 1.
Сравнительный анализ последовательностей аминокислот. а – на участке белка Тbх5 человека, содержащего замену p.Gly238Ala, с некоторыми животными; б – с другими T-box-содержащими белками у человека.
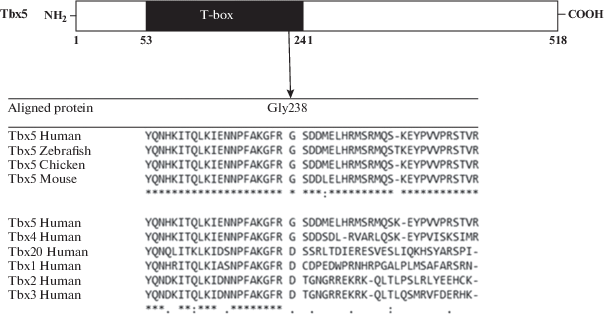
Между гистидином и аланином существует небольшая физико-химическая разница. Анализ функциональной значимости замены с использованием инструментов in silico (PolyPhen2, SIFT, Mutation tester) показал, что данный вариант вероятно повреждает структуру и/или функцию белка. В базе ClinVar представлен расположенный рядом вариант неопределенной значимости c.712G>A (VUS – variant of uncertain significance), в результате которого происходит замена этой же аминокислоты на серин (p.Gly238Ser, rs1593866534). Кроме того, соседний 237 кодон является “горячей точкой” нуклеотидных замен и к настоящему моменту в нем описаны три патогенные мутации: c.710G>C (p.Arg237Pro, rs104894378), c.710G>A (p.Arg237Gln, rs104894378), c.709C>T (p.Arg237Trp, rs104894382), что подтверждает функциональную важность данной области. Перечисленные варианты, за исключением p.Arg237Gln и p.Arg237Pro, не наблюдались ни в одном из известных проектов секвенирования экзома, что указывает на их диагностическую значимость. Встречаемость минорного аллеля (C>G/T в 710 положении нуклеотидной последовательности) в случае мутаций p.Arg237Gln и p.Arg237Pro также является очень низкой и варьирует в пределах 0.0005–0.000008 и, следовательно, они также не являются распространенными доброкачественными заменами в обследованных популяциях.
Функциональные исследования демонстрируют, что перечисленные мутации в гене TBX5 вызывают снижение ДНК-связывающей способности, в результате чего уменьшается взаимодействие фактора транскрипции Tbx5 с гомеобокс-содержащими факторами транскрипции NKX2-5 и GATA4 [22, 23], контролирующими формирование и развитие сердца. Имеющиеся данные о функциональной значимости аминокислот Arg237 и Gly238, с большой долей вероятности позволяют утверждать, что другие миссенс-мутации в этих кодонах, включая выявленную в данном исследовании, также будут неблагоприятно сказываться на работе белка Tbx5.
Следует отметить, что окончательный диагноз обследуемой нами пациентки был поставлен только после проведения генетического тестирования и обнаружения мутации в гене TBX5, поскольку клинический фенотип заболевания не удовлетворял всем характеристикам HOS. В ходе тщательного дополнительного обследования установлены некоторые скелетные аномалии, включающие деформацию грудного отдела позвоночника (сколиоз) и дисплазию лопатки. Так, например, у некоторых пациентов с HOS описан характерный вид узких наклонных плеч из-за сочетания коротких ключиц, гипоплазии головки плечевой кости и уменьшения мускулатуры [24].
Выявленные при ЭхоКГ-исследовании признаки недостаточности митрального и трикуспидального клапанов с регургитацией I–II степени, а также истончение межпредсердной перегородки также свидетельствовали в пользу патогенности выявленной новой мутации в гене TBX5. Одним из наиболее значимых проявлений заболевания у данной пациентки являлось наличие пароксизмов желудочковой тахикардии/фибрилляции желудочков с рецидивирующими синкопальными состояниями в молодом возрасте. В 42 года при внезапно возникшем синкопальном состоянии был зафиксирован эпизод ЖТ/ФЖ, потребовавший проведения реанимационных мероприятий и имплантации ИКД. Злокачественные нарушения ритма описываются у пациентов с HOS [2, 3] преимущественно при наличии ВПС в результате гемодинамических эффектов. В данном случае пароксизмы ЖТ/ФЖ были зарегистрированы у пациентки при отсутствии ВПС, дилатации камер сердца и гемодинамической перегрузки. Возникновению жизнеугрожающих аритмий и синкопе могли способствовать и обнаруженные у пациентки дополнительные редкие варианты в генах SCN5A и ANK2 (табл. 1), кодирующих альфа-субъединицу сердечного натриевого канала Nav1.5 и регуляторный белок анкирин-В соответственно. Мутации в обоих генах приводят к нарушению функционирования натриевого ионного канала и развитию жизнеугрожающих аритмий.
Таблица 1.
Дополнительные мутации у пациентки А. с синдромом Холта–Орама
| Ген | Хромосома (экзон) | Нуклеотидная замена/rs | Аминокислотная замена | Статус мутации | Частота минорного аллеля в популяциях (MAF) |
|---|---|---|---|---|---|
| SCN5A | 3 (20) | c.35758G>A/rs41261344 | p.Arg1193Gln | B/LB | 0.0008–0.07 |
| ANK2 | 4 (42) | c.10901T>A/rs66785829 | p.Val3634Asp | B/LB/VUS | 0.0009–0.01 |
Нуклеотидный вариант c.35758G>A в гене SCN5A является хорошо известной редкой мутацией (в среднем, значение MAF < 0.01), которая приводит к аминокислотной замене p.Arg1193Gln в α-субъединице натриевого канала. Данная замена имеет несколько повышенную встречаемость в восточноазиатских популяциях (MAF = 0.07; Exome Aggregation Consortium). Существующие клинические и эпидемиологические исследования в отношении этого варианта демонстрируют несколько противоречивые результаты [25]. Он идентифицирован у пациентов с LQT3 и BrS, а также у лиц с внезапной сердечной смертью (ВСС) [26, 27]. Сообщалось также, что данная мутация дестабилизирует инактивацию каналов и может являться фактором риска перечисленных синдромов [28]. В то же время, не выявлено статистически значимых различий по частоте минорной аллели между группами пациентов с аритмией, у большинства из которых были структурные заболевания сердца, и здоровым контролем в Японии (0.063 для обеих групп), между случаями с синдромом ВСС и контрольной группой на юге Китая (0.0608 и 0.0476 соответственно), а также между пациентами с полным блокадой атриовентрикулярной проводимости и контрольной группой в Корее (0.071 и 0.082 соответственно), что указывает на вероятную доброкачественность этой замены [29, 30]. Можно предположить, что влияние аллельного варианта c.35758G>A в гене SCN5A на формировании клинического фенотипа проявляется при наличии определенных дополнительных факторов, в том числе генетических. В данном случае таким фактором может быть выявленный редкий вариант c.10901T>A (p.Val3634Asp) в гене ANK2 с противоречивой оценкой патогенной значимости (табл. 1) и значением MAF < 0.01. Как известно, анкирин-В в качестве мембранного “адаптера” связывается с различными белками и участвует в регуляции потенциал-зависимого натриевого канала Nav1.5.
ЗАКЛЮЧЕНИЕ
В ходе проведенного генетического тестирования пациентки с мягким фенотипическим проявлением синдрома HOS и злокачественными желудочковыми тахиаритмиями (желудочковая тахикардия и фибрилляция желудочков) выявлена новая мутация c.713G>C (p.Gly238Ala) в экзоне 7 гена TBX5, а также два редких нуклеотидных варианта p.Val3634Asp (rs66785829) в гене ANK2 и p.Arg1193Gln (rs41261344) в гене SCN5A.
В представленном случае основной причиной наблюдаемого клинического фенотипа, по всей видимости, являлась новая мутация в гене TBX5. Вероятно патогеннная значимость этого нуклеотидного варианта подтверждается данными анализа с использованием предикторов in silico (PolyPhen2, SIFT, Mutation tester), а также ее локализацией в “горячей точке” функционально-значимых мутаций и фенотипическими проявлениями, характерными для синдрома HOS. Можно предположить, что нуклеотидные замены в генах SCN5A и ANK2 вносят определенный вклад в более тяжелое течение заболевания в виде жизнеугрожающих аритмий, демонстрируя аддитивный эффект трех мутаций. Однако данное предположение требует дополнительного изучения.
Приведенный клинический случай показывает важность генетического тестирования для установления точного диагноза у пациентов, имеющих нарушения опорно-двигательной системы верхних конечностей, ВПС и нарушения ритма.
Работа выполнена в рамках мероприятия 254 “Разработать метод диагностики наследственных нарушений сердечного ритма и/или проводимости c высоким риском внезапной сердечной смерти” подпрограммы 1 “Инновационные биотехнологии 2020” ГП “Наукоемкие технологии и техника”, 2016–2020 гг.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Greulich F., Rudat C., Kispert A. Mechanisms of T-box gene function in the developing heart // Cardiovasc. Res. 2011. V. 91. № 2. P. 212–222. https://doi.org/10.1093/cvr/cvr112
Stennard F.A., Harvey R.P. T-box transcription factors and their roles in regulatory hierarchies in the developing heart // Development. 2005. V. 132. P. 4897–4910. https://doi.org/0.1242/dev.02099
Hatcher C.J., Goldstein M.M., Mah C.S. et al. Identification and localization of TBX5 transcription factor during human cardiac morphogenesis // Dev. Dyn. 2000. V. 219. № 1. P. 90–95. https://doi.org/10.1002/1097-0177(200009)219:1<90::AID-DVDY1033>3.0.CO;2-L
Postma A.V., Christoffels V.M., Bezzina C.R. Developmental aspects of cardiac arrhythmogenesis // Cardiovasc. Res. 2011. V. 91. № 2. P. 243–251. https://doi.org/10.1093/cvr/cvr134
Packham E.A., Brook J.D. T-box genes in human disorders // Hum. Mol. Genet. 2003. V. 12. № 1. P. 37–44. https://doi.org/10.1093/hmg/ddg077
Spiridon M.R., Petris A.O., Gorduza E.V. et al. Holt-Oram Syndrome with multiple cardiac abnormalities // Cardiol. Res. 2018. V. 9. № 5. P. 324–329. https://doi.org/10.14740/cr767w
Basson C.T., Huang T., Lin R.C. et al. Different TBX5 interactions in heart and limb defined by Holt–Oram syndrome mutations // PNAS. 1999. V. 96. № 6. P. 2919–2924. https://doi.org/10.1073/pnas.96.6.2919
Holt M., Oram S. Familial heart disease with skeletal malformations // Br. Heart. J. 1960. V. 22. № 2. P. 236–242. https://doi.org/10.1136/hrt.22.2.236
Prevalence and incidence of rare diseases: Bibliographic data // Orphanet Rep. Series. 2020. № 1. P. 1–78. http://www.orpha.net/orphacom/cahiers/docs/GB/ Prevalence_of_rare_diseases_by_alphabetical_list.pdf.
Basson C.T., Bachinsky D.R., Lin R.C. et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome // Nat. Genet. 1997. V. 15. № 1. P. 30–35. https://doi.org/10.1038/ng0197-30
Li Q.Y., Newbury-Ecob R.A, Terrett J.A. et al. Holt–Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family // Nat. Genet. 1997. V. 15. P. 21–29. https://doi.org/10.1038/ng0197-21
Zhu T., Qiao L., Wang Q. et al. T-box family of transcription factor-TBX5, insights in development and disease // Am. J. Transl. Res. 2017. V. 9. № 2. P. 442–453.
Vanlerberghe C., Jourdain A.S., Ghoumid J. et al. Holt-Oram syndrome: clinical and molecular description of 78 patients with TBX5 variants // Eur. J. Hum. Genet. 2019. V. 27. № 3. P. 360–368. https://doi.org/10.1038/s41431-018-0303-3
Brassington A.M., Sung S.S., Toydemir R.M. et al. Expressivity of Holt-Oram syndrome isnot predicted by TBX5 genotype // Am. J. Hum. Genet. 2003. V. 73. № 1. P. 74–85. https://doi.org/10.1086/376436
McDermott D.A., Bressan M.C., He J. et al. TBX5 genetic testing validates strict clinical criteria for Holt–Oram syndrome // Pediatr. Res. 2005. V. 58. № 5. P. 981–986. https://doi.org/10.1203/01.PDR.0000182593.95441.64
Debeer P., Race V., Gewillig M. et al. Novel TBX5 mutations in patients with Holt–Oram syndrome // Clin. Orthop. Relat. Res. 2007. V. 462. P. 20–26.https://doi.org/10.1097/BLO.0b013e3181123ffe
Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data // Nucl. Ac. Res. 2010. V. 38. № 16. P. e164. https://doi.org/10.1093/nar/gkq603
Adzhubei I.A., Schmidt S., Peshkin L. et al. A method and server for predicting damaging missense mutations // Nat. Methods. 2010. V. 7. № 4. P. 248–249. https://doi.org/10.1038/nmeth0410-248
Kumar P., Henikoff S., Nag P.C. Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm // Nat. Protoc. 2009. V. 4. № 7. P. 1073–1081. https://doi.org/10.1038/nprot.2009.86
Shihab H.A., Gough J., Cooper D.N. et al. Predicting the functional, molecular and phenotypic consequences of amino acid substitutions using hidden Markov models // Hum.Mutat. 2013. V. 34. № 1. P. 57–65. https://doi.org/10.1002/humu.22225
Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. Mutation Taster evaluates disease-causing potential of sequence alterations // Nat. Methods. 2010. V. 7. № 8. P. 575–576. https://doi.org/10.1038/nmeth0810-575
Kimura M., Kikuchi A., IchinoiN., Kure S. Novel TBX5 duplication in a Japanese family with Holt-Oram syndrome // Pediatr. Cardiol. 2015. V. 36. № 1. P. 244–247. https://doi.org/10.1007/s00246-014-1028-x
Dreßen M., Lahm H., Lahm A. et al. A novel de novo TBX5 mutation in a patient with Holt-Oram syndrome leading to a dramatically reduced biological function // Mol. Genet. Genomic Med. 2016. V. 4. № 5. P. 557–567. https://doi.org/10.1002/mgg3.234
Smith A.T., Sack G.H., Jr., Taylor G.J. Holt-Oram syndrome // J. Pediatr. 1979. V. 95. № 4. P. 538–543. https://doi.org/10.1016/s0022-3476(79)80758-1
Abe M., Kinoshita K., Matsuoka K. et al. Lack of modulatory effect of the SCN5A R1193Q polymorphism on cardiac fast Na+ current at body temperature // PLoS One. 2018. V. 13. № 11. P.: e0207437. https://doi.org/10.1371/journal.pone.0207437
Matsusue A., Kashiwagi M., Hara K. et al. An autopsy case of sudden unexpected nocturnal death syndrome with R1193Q polymorphism in the SCN5A gene // Legal Med. (Tokyo). 2012. V. 14. № 6. P. 317–319. https://doi.org/10.1016/j.legalmed.2012.04.009
Hwang H.W., Chen J.J., Lin Y.J. et al. R1193Q of SCN5A, a Brugada and long QT mutation, is a common polymorphism in Han Chinese // J. Med. Genet. 2005. V. 42. № 2. P.: e7. https://doi.org/10.1136/jmg.2004.027995
Maekawa K., Saito Y., Ozawa S. et al. Genetic polymorphisms and haplotypes of the human cardiac sodium channel alpha subunit gene (SCN5A) in Japanese and their association with arrhythmia // Ann. Hum. Gen. 2005. V. 69. № 4. P. 413–428. https://doi.org/10.1046/j.1529-8817.2005.00167.x
Cheng J., Makielski J.C., Yuan P. et al. Sudden unexplained nocturnal death syndrome in Southern China: An epidemiological survey and SCN5A gene screening // Am. J. Forensic Med. Pathol. 2011. V. 32. № 4. P. 359–363. https://doi.org/10.1097/PAF.0b013e3181d03d02
Park H.S., Kim Y.N., Lee Y.S. et al. Genetic analysis of SCN5A in Korean patients associated with atrioventricular conduction block // Genomics Inform. 2012. V. 10. № 2. P. 110–116. https://doi.org/10.5808/GI.2012.10.2.110
Дополнительные материалы отсутствуют.