

Генетика, 2022, T. 58, № 5, стр. 581-590
Метилирование гена МАРТ при нейродегенеративных заболеваниях из группы синуклеинопатий
Е. В. Яковенко 1, *, Н. Ю. Абрамычева 1, Е. Ю. Федотова 1, С. Н. Иллариошкин 1
1 Научный центр неврологии
125367 Москва, Россия
* E-mail: helenaabracham@gmail.com
Поступила в редакцию 21.10.2021
После доработки 09.12.2021
Принята к публикации 27.12.2021
- EDN: QXVFGU
- DOI: 10.31857/S0016675822050113
Аннотация
К группе синуклеинопатий – нейродегенеративных заболеваний, связанных с накоплением в структурах центральной нервной системы патологических белковых агрегатов белка альфа-синуклеина – относятся такие заболевания как болезнь Паркинсона (БП) и мультисистемная атрофия (МСА). Точные причины развития этих заболеваний на настоящий момент еще не определены, при этом установлено взаимодействие генетических и средовых факторов. Полиморфные варианты гена МАРТ, а также нарушение эпигенетических механизмов, в частности, метилирования транскрипционно значимых областей генов, являются факторами риска развития синуклеинопатий. В работе изучено влияние уровня метилирования гена МАРТ на развитие БП и МСА, проведено клинико-эпигенетическое сопоставление данных. Определено гиперметилирование в трех CpG-сайтах промоторной области гена МАРТ в группе МСА при сравнении с контролем. Также определена статистически значимая разница между гаплотипами МАРТ в уровне метилирования четырех CpG-сайтов промотора в группе МСА, при этом протективный гаплотип Н2 имел более низкие уровни метилирования. Определено влияние возраста и противопаркинсонической терапии агонистами дофаминовых рецепторов на уровень метилирования гена МАРТ. Наши данные сравнительного анализа метилирования при синуклеинопатиях свидетельствуют скорее о возможной протективной роли гипометилирования МАРТ, на что указывают преимущественно гипометилированный статус СрG-сайтов в контрольной группе, гипометилирование протективного Н2-гаплотипа МАРТ и повышение уровня гиперметилирования с возрастом.
Синуклеинопатии представляют собой одну из групп церебральных протеинопатий – нейродегенеративных заболеваний, характеризующихся накоплением в нейронах и глиальных клетках патологических белковых агрегатов [1]. При синуклеинопатиях таким патологическим субстратом являются агрегаты короткого синаптического белка альфа-синуклеина, принимающего аномальную конформацию, в структурах центральной и периферической нервной системы [2]. Различия в типах клеток и областях нервной системы, где локализуются агрегаты, обусловливают клинические проявления синуклеинопатий и манифестацию их различных нозологических форм, среди которых наиболее распространенными являются болезнь Паркинсона (БП) и мультисистемная атрофия (МСА).
Согласно эпидемиологическим исследованиям последних лет БП является глобальным бременем для здравоохранения и общества в целом, поражая 1–2% населения старше 65 лет и 4–5% людей старше 85 лет [3, 4]. Около 10% случаев заболевания затрагивает более молодую возрастную группу до 50 лет [5, 6]. Проявляется БП как моторными симптомами (гипокинезия, тремор, ригидность, постуральная неустойчивость), так и широким рядом немоторных нарушений [7]. Главным патоморфологическим признаком этого заболевания являются агрегаты белка альфа-синуклеина в цитоплазме нейронов – тельца Леви, которые определяются в комбинации с потерей дофаминергических нейронов компактной части черной субстанции среднего мозга [8].
БП – комплексная патология, которая вызывается сочетанием генетических и средовых факторов, причем на долю спорадической формы приходится около 90%, а на долю наследственных форм – около 10% всех случаев заболевания. На данный момент известны 23 гена (SNCA, PARK2, LRRK2, PINK1, DJ-1, GBA и др.), мутации в которых вызывают развитие наследственных форм БП [9]. Также известно множество полиморфных вариантов в различных генах (включая SNCA и МАРТ), ассоциированных с БП и установленных по результатам полногеномных ассоциативных исследований – GWAS (genome wide association study) [10–12].
МСА – прогрессирующее нейродегенеративное заболевание, характеризующееся вегетативной недостаточностью в комбинации с моторными нарушениями, преимущественно паркинсонического типа и/или мозжечкового типа [13]. При МСА агрегаты альфа-синуклеина встречаются главным образом в виде олигодендроглиальных цитоплазматических включений и, в меньшей степени, нейрональных цитоплазматических включений, что приводит к дегенерации клеток различных областей мозга [14]. МСА считается спорадическим заболеванием, при этом, по данным последних GWAS-исследований, обнаружено несколько локусов, ассоциированных с риском развития МСА – в их числе гены SNCA, MAPT и COQ2 [15].
Поиск факторов предрасположенности к развитию синуклеинопатий ведется в различных направлениях, в том числе и в области эпигенетики. Эпигенетические изменения относятся к устойчивым и наследуемым изменениям в экспрессии генов, происходящих без нарушения последовательности ДНК [16, 17]. К основным эпигенетическим механизмам относят метилирование ДНК, модификации гистонов и контроль активности генов с помощью некодирующих РНК [18]. Эпигенетические изменения, влияющие на развитие БП, активно изучаются в течение последнего десятилетия и сконцентрированы на метилировании ДНК – процессе присоединения метильной группы к цитозину, состоящему в тандеме с гуанином (CpG-динуклеотид, или CpG-сайт). Считается, что высокий уровень метилирования CpG-сайтов в промоторных и регуляторных областях приводит к нарушению связывания с ДНК-полимеразами и транскрипционными факторами и, соответственно, к снижению транскрипции генов, а низкий – наоборот – к повышению транскрипции [19].
Учитывая подтвержденную во многих исследованиях тесную взаимосвязь между уровнем экспрессии альфа-синуклеина и риском развития БП [20–23], метилирование альфа-синуклеина стало наиболее изучаемым аспектом при оценке роли эпигенетических механизмов у пациентов с БП. Известно, что в гене альфа-синуклеина (SNCA) существуют CpG-островки (области с высокой концентрацией CpG-динуклеотидов), находящиеся в промоторной области и в интроне 1, причем по данным ряда проведенных исследований CpG-сайты этой области SNCA гипометилированы в группе пациентов с БП, что приводит к повышению синтеза мРНК SNCA и накоплению альфа-синуклеина в клетках [24–29].
Помимо гена SNCA многие другие гены также показали значимую роль в формировании предрасположенности к развитию БП и других синуклеинопатий. Ген MAPT, который кодирует тау-белок, ассоциированный с микротрубочками цитоскелета нейронов, согласно ряду GWAS-исследований является фактором риска развития как БП, так и МСА [30, 31]. Обнаружено, что гаплотип Н1 гена МАРТ повышает риск развития БП, в то время как гаплотип Н2 является протективным фактором в отношении этого заболевания [32]. Исследований, посвященных изучению влияния метилирования гена МАРТ на развитие БП и МСА, на настоящий момент крайне мало. В гене МАРТ расположено несколько CpG-островков, причем самый большой из них, содержащий 302 CpG-сайта, располагается в промоторном регионе [33].
Целью нашего исследования стало изучение влияния уровня метилирования гена МАРТ на развитие БП и МСА, проведение клинико-эпигенетических сопоставлений, а также изучение корреляций между гаплотипами МАРТ и уровнем метилирования.
МАТЕРИАЛЫ И МЕТОДЫ
Пациенты
В исследование включены 61 пациент с диагнозом “болезнь Паркинсона”, поставленным по критериям Международного общества расстройств движений (32 мужчины, 29 женщин, средний возраст 60.4 лет, медиана 63 [53; 69 ] лет). В группу МСА вошли 22 пациента с паркинсоническим фенотипом данного заболевания, поставленным по критериям S. Gilman с соавт. [34]: 7 мужчин, 15 женщин, средний возраст 61.0 лет, медиана 59.5 [54.5; 69 ] лет. В группу контроля включены 43 неврологически здоровых добровольца: 16 мужчин, 27 женщин, средний возраст – 58.0 [53; 62.5 ] лет. Группы были сопоставимы по поло-возрастным характеристикам.
Всем пациентам с БП проводился подробный сбор анамнеза, общеклинический и неврологический осмотр, уточнялись возраст дебюта и длительность заболевания, семейный анамнез, определялись стадия заболевания по Hoehn-Yahr, форма заболевания (дрожательная/акинетико-ригидная/смешанная), а также проводилось тестирование пациентов по Унифицированной шкале оценки болезни Паркинсона Международного общества расстройств движений (MDS-UPDRS), Монреальской шкале оценки когнитивных функций (МОСА), Госпитальной шкале тревоги и депрессии (HADS).
Средний возраст дебюта заболевания в группе БП составлял 55 лет [48; 64 ] лет. Длительность заболевания составляла в среднем 4 [3; 7] года. В группе преобладали пациенты со смешанной (дрожательно-ригидной) формой БП – 49 (80.3%) больных, акинетико-ригидную форму определили у 11 (18.0%) пациентов, дрожательная форма была у одного пациента (1.6%). В состав основной группы БП вошли 13 пациентов (21.3%) с положительным семейным анамнезом по данному заболеванию.
Пациенты были подразделены на следующие стадии по функциональной шкале Hoehn-Yahr: 1‑я стадия – 12 человек (19.9%), 2-я стадия – 18 человек (29.5%), 2.5-я стадия – 1 человек (1.6%), 3-я стадия – 28 человек (46.0%), 4-я стадия – 2 человека (3.3%). Средний суммарный балл по шкале UPDRS в периоде выключения был равен 55 [35; 78.75] баллов.
Противопаркинсоническую терапию получал 41 пациент (67.2%). При этом препараты леводопы принимал 31 (50.8%) пациент, средняя суточная доза составила 600 [300; 737.25] мг. Терапию агонистами дофаминовых рецепторов (прамипексол, ропинирол, пирибедил) получали 30 пациентов (49.2%), при этом средняя эквивалентная доза составила 200 [150; 300] мг. Амантадины получали 20 пациентов (32.79%), со средней эквивалентной дозой 300 [150; 300] мг. Также рассчитывалась суммарная эквивалентная доза в подгруппе пациентов (n = 41), получающих терапию, которая составила 600 [300; 950] мг. При расчете в общей группе пациентов (n = 61) суммарная эквивалентная доза принимаемых препаратов составила 300 [0; 800] мг.
В группе МСА средний возраст начала болезни составил 55 [52.25; 64 ] года, длительность заболевания – 3.5 [2; 5] года.
Методы исследования
В гене МАРТ уровень метилирования определялся в промоторной области (CpG 11–29, нумерация CpG-сайтов от начала исследуемого CpG-островка, охватывающего также нетранслируемый 1-й экзон) (рис. 1). Нумерация экзонов проведена согласно референсному транскрипту NM_001123066.4. Расположение исследованной области относительно референсного генома (GRCh38): chr17: 45 894 045 (CpG-11) – chr17: 45 894 235 (CpG-29). CpG-сайты с 1-го по 10-й в экзоне 1 были полностью метилированы в обеих исследованных группах, поэтому в дальнейшем они не включались в анализ.
Рис. 1.
Локализация исследуемой промоторной области с экзоном 1, и варианта rs1052553 в экзоне 11 гена МАРТ (синие элементы – облигатно транслируемые экзоны, красный, зеленый, желтый – экзоны с альтернативным сплайсингом в тканях головного мозга и других тканях, фиолетовые – экзоны, не транслируемые в головном мозге, белые – нетранслируемые области). Зеленый овал – промоторная область гена МАРТ. Буквенная последовательность – изучаемые CpG-сайты гена МАРТ (обратный праймер отмечен зеленым цветом).

Подобранные праймеры для исследования уровня метилирования экзона 1 гена МАРТ:
• прямой праймер 5'→3' TGTTAAGGAAAGGATTTATTTTGGTT,
• обратный праймер 5'→3' CTTTCTCCACCTCCTATAATTAAAATCT.
Паттерн метилирования определялся с помощью метода прямого секвенирования амплифицированных участков ДНК после выделения геномной ДНК из лейкоцитов периферической крови и ее бисульфитной конверсии набором EZ DNA Methylation Kit (Zymo Research, США). Амплификацию фрагментов ДНК для последующего сиквенса проводили в 10 мкл реакционной среды: 50 мM KCl, 50 мM Трис-HСl (pH 8.8), 2.5 мМ MgCl2, 250 мкМ dNTP, 1 ед. Taq ДНК-полимеразы с ингибирующими активность фермента антителами (“Синтол”, Москва), по 1 мкМ прямого и обратного праймеров, образец ДНК ~20 нг.
Визуализация осуществлялась с помощью программного обеспечения Sequencing Analysis Software (v5.2 Applied Biosystems). Степень метилирования рассчитывали путем анализа первичных результатов секвенирования по Сэнгеру. Процент метилирования для каждого конкретного CpG-сайта для каждого ДНК-образца рассчитывали по отношению высоты синего пика С (пик электрофореграммы, местоположение которого соответствует анализируемому CpG-сайту и указывающий на наличие метилированного цитозина) относительно суммарной высоты пиков С + Т данного положения (метилированный синий и неметилированный красный цитозин).
Гаплотип гена МАРТ определялся с помощью сцепленного полиморфного варианта rs1052553 (A>G) (где нуклеотид А соответствует гаплотипу Н1, а нуклеотид G – гаплотипу Н2) в экзоне 11 (рис. 1). Амплификацию фрагментов двухцепочечной ДНК для последующего сиквенса проводили в 10 мкл специально подобранной реакционной среды: 50 мM KCl, 50 мM Трис-HСl (pH 8.8), 2.5 мМ MgCl2, 250 мкМ dNTP, 1 ед. Taq ДНК-полимеразы с ингибирующими активность фермента антителами (“Синтол”, Москва), по 1 мкМ прямого и обратного праймера, образец ДНК ~20 нг.
Подобранные праймеры для экзона 11 гена МАРТ:
• прямой праймер 5'→3' AAGACTGTGGAGCCGAGTTG,
• обратный праймер 5'→3' TGCCCTGACTATGAGAGCCT.
Статистический анализ проводился на программе Statistica 13 (Tibco Russia). В работе использовались: U-критерий Манна–Уитни для оценки различий между двумя независимыми выборками, ANCOVA, метод ранговой корреляции Спирмена и множественная линейная регрессия. Статистический уровень значимости принимался равным 0.05. Также проводилась поправка на множественную проверку гипотез по методу Бонферрони, для 19 CpG-сайтов, р составило 0.0026.
РЕЗУЛЬТАТЫ
Метилирование МАРТ у пациентов с БП и МСА
При сравнении групп БП и контроля не было обнаружено статистически значимых различий в уровнях метилирования изучаемых сайтов в гене МАРТ.
При сравнении групп МСА и контроля были обнаружены статистические значимые различия в уровнях метилирования ряда CpG-сайтов, представленных в табл. 1. Уровень метилирования MAPT в группе БП, МСА и в контрольной группе отражен на рис. 2. Как видно из рисунка, уровень метилирования при БП занимает промежуточное положение между пациентами с МСА и контрольной группой.
Рис. 2.
Метилирование CpG-сайтов промоторной области гена МАРТ. * – статистически значимые различия между контрольной группой и пациентами с МСА. По оси абсцисс – номера СpG-сайтов, по оси ординат – процент метилирования. БП – болезнь Паркинсона, МСА-П – мультисистемная атрофия – паркинсонический фенотип, К – группа контроля.
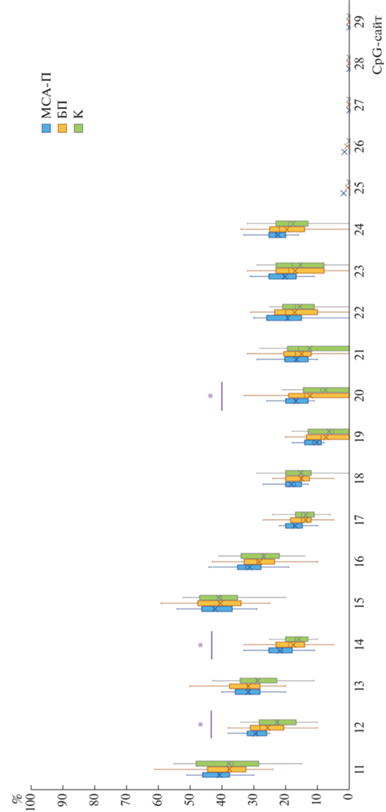
При сравнении уровня метилирования гена МАРТ в группах БП и МСА статистически значимой разницы в исследуемых СрG выявить не удалось.
Взамимосвязь метилирования МАРТ и гаплотипов Н1 и Н2
Процент встречаемости аллелей и диплотипов H1/H2 гена MAPT по группам приведен в табл. 2.
Таблица 2.
Распределение аллельных вариантов Н1 и Н2 по группам
| Группа | Гаплотип | Генотип | |||
|---|---|---|---|---|---|
| Н1 | Н2 | Н1/Н1 | Н1/Н2 | Н2/Н2 | |
| БП | 67 (88%) | 9 (12%) | 32 (84%) | 3 (8%) | 3 (8%) |
| МСА | 38 (82.6%) | 8 (17.4%) | 16 (69.5%) | 6 (26%) | 1 (4.5%) |
| Контроль | 36 (90%) | 4 (10%) | 17 (85%) | 2 (10%) | 1 (5%) |
Поочередное сравнение контрольной группы с группой БП и с группой МСА по распределению аллелей и генотипов отличий не выявило. Учитывая низкую частоту встречаемости диплотипов Н1/Н2 и Н2/Н2, анализ подгрупп диплотипов и их связи с метилированием не проводился.
Исследован уровень метилирования у пациентов с БП, МСА и в контрольной группе в зависимости от гаплотипа МАРТ (Н1 или Н2).
При сравнении метилирования у пациентов с БП, являющихся носителями разных гаплотипов, был обнаружен один дифференциально метилированный CpG-сайт – CpG-15: в группе пациентов с аллелем Н1 – метилирование составило 40 [34; 45 ] %, в группе с аллелем Н2 – 34 [30; 34]% (p(U) = 0.001819), т.е. носители Н1 имели более высокий уровень метилирования.
В группе пациентов с МСА также выявлены значимые различия в метилировании гена МАРТ при носительстве гаплотипов Н1 и Н2. Результаты приведены в табл. 3. Во всех четырех дифференциально метилированных сайтах носительство гаплотипа Н1 было ассоциировано с более высоким уровнем метилирования.
Таблица 3.
Метилирование сайтов (%) промоторной области гена МАРТ в зависимости от носительства аллелей Н1 и Н2 у пациентов с МСА
В контрольной группе различий между носителями гаплотипов выявлено не было.
Анализ метилирования МАРТ в зависимости от демографических и клинических характеристик
Различий в метилировании МАРТ между мужчинами и женщинами выявлено не было.
Изучена взаимосвязь между уровнем метилирования МАРТ и возрастом пациентов на момент исследования. В группе БП были выявлены прямые корреляции указанных показателей в трех CpG-сайтах: CpG-19 (r = 0.52 – средняя корреляция, р = 0.000001); CpG-22 (r = 0.47 – слабая корреляция, р = 0.00013); CpG-23 (r = 0.4 – слабая корреляция, р = 0.00173). Таким образом, с возрастом наблюдалось гиперметилирование исследованных сайтов. При этом в группе пациентов с МСА и в контрольной группе подобных корреляций обнаружить не удалось.
Корреляций между возрастом начала заболеваний (с поправкой на возраст на момент обследования) и уровнем метилирования в МАРТ в работе не обнаружено.
При изучении корреляций уровня метилирования МАРТ с длительностью заболевания в группе БП выявлена слабая обратная корреляция по сайту CpG-15 (r = –0.38, р = 0.00259). В группе МСА корреляций с длительностью заболевания не выявлено.
При исследовании уровня метилирования пациентов с БП в зависимости формы заболевания (акинетико-ригидная/дрожательная/смешанная) и семейного анамнеза (отягощен/не отягощен) статистически значимых различий выявлено не было. При оценке корреляций между уровнем метилирования и стадией заболевания по шкале Hoehn-Yahr, суммой баллов по шкале UPDRS и ее подразделам, а также суммой баллов по шкалам HADS и МОСА (с поправкой на возраст) значимых корреляций выявлено не было.
Согласно полученным данным, терапия леводопой или амантадинами не влияла на профиль метилирования MAPT. В то же время обнаружена разница между группами пациентов с БП, принимающими и непринимающими агонисты дофаминовых рецепторов. При поправке на возраст (которая была сделана с учетом того, что агонисты дофаминовых рецепторов обычно назначаются более молодым пациентам) выявлены различия по следующим сайтам МАРТ: CpG-22 (без терапии – 22 [19; 26]%, с терапией – 11 [8; 20.25]%, р = 0.000178) и CpG-23 (без терапии – 21 [18; 25]%, с терапией – 10.5 [6; 21.25]%, р = = 0.001689), то есть назначение агонистов снижает уровень метилирования.
Корреляций между уровнем метилирования и дозой леводопы, дозой агонистов дофаминовых рецепторов (с поправкой на сумму баллов по шкале МОСА и возраст), эквивалентной дозой всех препаратов не выявлено.
ОБСУЖДЕНИЕ
В настоящей работе нами был исследован ген МАРТ, роль которого в риске развития нейродегенеративных заболеваний из группы синуклеинопатий и таупатий была установлена в предыдущих работах. CpG-островок промоторной области с экзоном 1 был выбран в связи с тем, что именно этот участок гена МАРТ определяет его экспрессию.
Нами при анализе эпигенетических модификаций МАРТ определено гиперметилирование в трех промоторных CpG-сайтах в группе МСА при сравнении с контролем. При этом различий между пациентами с БП и контрольной группой, а также между группами БП и МСА выявлено не было.
Одно из небольшого числа исследований по метилированию промотора МАРТ при БП, проведенное K. Coupland с соавт. [35], показало разницу между БП и контролем: у пациентов с БП ген МАРТ был гиперметилирован в мозжечке (регионе, обычно не вовлеченном в патологический процесс при БП) и гипометилирован в скорлупе (регионе, в котором наиболее выражены патологические изменения при БП). Авторы предположили, что гиперметилирование промотора МАРТ может выполнять протективную функцию, понижая экспрессию МАРТ. Такое предположение частично подтверждается и данными исследования, проведенного при болезни Альцгеймера: у пациентов определялось гипометилирование CpG-островка промоторной области гена МАРТ. При этом предполагается, что гипометилирование МАРТ у пациентов с болезнью Альцгеймера ассоциировано с повышенной экспрессией тау-протеина и его патологической агрегацией, что наблюдается в мозге при данном заболевании [36].
Наши данные сравнительного анализа метилирования при синуклеинопатиях свидетельствуют скорее о возможной протективной роли гипометилирования МАРТ. На это указывают преимущественно гипометилированный статус СрG-сайтов в контрольной группе, гипометилирование протективного Н2-гаплотипа МАРТ и повышение уровня гиперметилирования с возрастом (возраст – один из основных факторов риска развития синуклеинопатий). Возможно, что характер метилирования МАРТ достаточно специфичен для разных видов нейродегенеративной патологии [37].
Ассоциация между БП и вариантами в гене MAPT активно изучается в различных популяциях, и наиболее значимыми считаются полиморфизмы, связанные с гаплотипом МАРТ (Н1 или Н2). Известно, что гаплотип Н1 и некоторые SNP в гене МАРТ повышают риск развития БП, а гаплотип H2, напротив, имеет протективные свойства. Такая зависимость позволила предположить, что эпигенетические модификации МАРТ могут влиять на развитие БП. В настоящей работе определялась зависимость уровня метилирования CpG-сайтов гена МАРТ от гаплотипа – Н1 или Н2. Следует отметить, что гаплотип Н2 достаточно редко встречается в российской популяции, поэтому носительство генотипов Н1/Н2 и, тем более, Н2/Н2, было слабо представлено среди нашей выборки пациентов и лиц контрольной группы. Тем не менее, нами была определена статистически значимая разница между гаплотипами в уровне метилирования четырех CpG-сайтов промоторной области гена МАРТ в группе МСА, при этом протективный гаплотип Н2 имел более низкие уровни метилирования.
K. Coupland с соавт. в группе пациентов с БП показали более высокий уровень метилирования CpG-сайтов в гене МАРТ у носителей гаплотипа Н1 в сравнении с гаплотипом Н2 [35]. Группой Y. Li с соавт. проведено исследование метилирования гена МАРТ при таупатиях и выявлено повышение уровня метилирования в клетках крови и в регионах головного мозга при гаплотипе Н1 в сравнении с гаплотипом Н2 [38]. Данные работы согласуются с нашими результатами.
В настоящей работе проводился анализ связи метилирования с возрастом пациентов на момент исследования, который продемонстрировал прямые корреляции c метилированием 3 CpG-сайтов промотора МАРТ в группе БП – по мере старения уровень метилирования повышался. При этом метилирование не было связано с возрастом начала заболевания при поправке на возраст. Статистически значимых различий в уровне метилирования CpG-сайтов между женщинами и мужчинами в группах БП, МСА и контроля обнаружено не было. Полученные результаты не согласуются с работой [35], в который было выявлено, что у женщин уровень метилирования был значимо выше, а возраст начала заболевания прямо коррелировал с метилированием MAPT.
В представленном исследовании были изучены корреляции между уровнями метилирования и различными клиническими проявлениями при БП и МСА. Нам не удалось выявить корреляций уровня метилирования ни с формой заболевания, ни с тяжестью моторных или немоторных клинических проявлений.
Также мы не обнаружили связи между дозой леводопы, эквивалентной дозой всех принимаемых противопаркинсонических препаратов, наличием терапии леводопой и амантадинами и профилем метилирования гена MAPT. В то же время нами обнаружено различие в профиле метилирования между пациентами, принимающими и не принимающими агонисты дофаминовых рецепторов: пациенты без терапии этой группой препаратов имели значимо более высокий уровень метилирования в трех промоторных CpG-сайтах гена МАРТ. Эти данные показывают, что терапия агонистами дофаминовых рецепторов может приводить к гипометилированию, что в свою очередь говорит о возможном модифицирующем воздействии дофаминовых агонистов на течение нейродегенеративного процесса посредством эпигенетических механизмов.
Выявленные изменения в паттернах метилирования МАРТ у пациентов с БП и МСА могут рассматриваться в качестве звеньев молекулярного патогенеза синуклеинопатий. Оценка уровня метилирования также может стать частью комплекса биомаркеров, которые исследуются в клинике для уточнения диагноза и прогноза болезни; согласно полученным нами данным, оценка метилирования MAPT может играть такую биомаркерную роль в первую очередь для МСА. Полученные данные свидетельствуют о возможном модифицирующем влиянии противопаркинсонической терапии на профиль метилирования и, следовательно, на течение заболевания, что требует дальнейших исследований.
Исследование проведено без спонсорской поддержки.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Kovacs G.G. Molecular pathological classification of neurodegenerative diseases: Turning towards precision medicine // Int. J. Mol. Sci. 2016. V. 17. № 2. 189. P. 1–33. https://doi.org/10.3390/ijms17020189
McCann H., Stevens C.H., Cartwright H., Halliday G.M. α-Synucleinopathy phenotypes // Parkinsonism Relat. Disord. 2014. V. 20. Suppl 1. P. 62–67. https://doi.org/10.1016/S1353-8020(13)70017-8
Bertram L., Tanzi R.E. The genetic epidemiology of neurodegenerative disease // J. Clin. Invest. 2005. V. 115. № 6. P. 1449–1457. https://doi.org/10.1172/JCI24761
Hirsch L., Jette N., Frolkis A. et al. The incidence of Parkinson’s disease: a systematic review and meta-analysis // Neuroepidemiology. 2016. V. 46. № 4. P. 292–300. https://doi.org/10.1159/000445751
Karimi-Moghadam A., Charsouei S., Bell B., Jabalameli M.R. Parkinson disease from mendelian forms to genetic susceptibility: New molecular insights into the neurodegeneration process // Cell. Mol. Neurobiol. 2018. V. 38. № 6. P. 1153–1178. https://doi.org/10.1007/s10571-018-0587-4
Schrag A., Ben-Shlomo Y., Quinn N.P. Cross sectional prevalence survey of idiopathic Parkinson’s disease and Parkinsonism in London // BMJ. 2000. V. 321. № 7252. P. 21–22. https://doi.org/10.1136/bmj.321.7252.21
Jankovic J. Parkinson’s disease: Clinical features and diagnosis // J. Neurol. Neurosurg. Psychiatry. 2008. V. 79. № 4. P. 368–376. https://doi.org/10.1136/jnnp.2007.131045
Dickson D.W. Parkinson’s disease and parkinsonism: neuropathology // Cold Spring Harb. Perspect. Med. 2012. V. 2. № 8. P. a009258. https://doi.org/10.1101/cshperspect.a009258
Jankovic J., Tan E.K. Parkinson’s disease: etiopathogenesis and treatment // J. Neurol. Neurosurg. Psychiatry. 2020. V. 91. № 8. P. 795–808. https://doi.org/10.1136/jnnp-2019-322338
Simón-Sánchez J., Schulte C., Bras J.M. et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease // Nat. Genet. 2009. V. 41. № 12. P. 1308–1312. https://doi.org/10.1038/ng.487
Nalls M.A., Pankratz N., Lill C.M. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease // Nat. Genet. 2014. V. 46. № 9. P. 989–993. https://doi.org/10.1038/ng.3043
Chang D., Nalls M.A., Hallgrímsdóttir I.B. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci // Nat. Genet. 2017. V. 49. № 10. P. 1511–1516. https://doi.org/10.1038/ng.3955
Ozawa T., Paviour D., Quinn N.P. et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: Clinicopathological correlations // Brain J. Neurol. 2004. V. 127. Pt. 12. P. 2657–2671. https://doi.org/10.1093/brain/awh303
Yoshida M. Multiple system atrophy: alpha-synuclein and neuronal degeneration // Neuropathology. 2007. V. 27. № 5. P. 484–493. https://doi.org/10.1111/j.1440-1789.2007.00841.x
Sailer A., Scholz S.W., Nalls M.A. et al. A genome-wide association study in multiple system atrophy // Neurology. 2016. V. 87. № 15. P. 1591–1598. https://doi.org/10.1212/WNL.0000000000003221
Portela A., Esteller M. Epigenetic modifications and human disease // Nat. Biotechnol. 2010. V. 28. № 10. P. 1057–1068. https://doi.org/10.1038/nbt.1685
Waddington C.H. The epigenotype. 1942 // Int. J. Epidemiol. 2012. V. 41. № 1. P. 10–13. https://doi.org/10.1093/ije/dyr184
Marques S., Oliveira C., Pereira C., Outeiro T. Epigenetics in neurodegeneration: A new layer of complexity // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011. V. 35. № 2. P. 348–355. https://doi.org/10.1016/j.pnpbp.2010.08.008
Wüllner U., Kaut O., deBoni L. et al. DNA methylation in Parkinson’s disease // J. Neurochem. 2016. V. 139. Suppl 1. P. 108–120. https://doi.org/10.1111/jnc.13646
Chartier-Harlin M.C., Kachergus J., Roumier C. et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease // Lancet. 2004. V. 364. № 9440. P. 1167–1169. https://doi.org/10.1016/S0140-6736(04)17103-1
Miller D.W., Hague S.M., Clarimon J. et al. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication // Neurology. 2004. V. 62. № 10. P. 1835–1838. https://doi.org/10.1212/01.wnl.0000127517.33208.f4
Nemani V.M., Lu W., Berge V. et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis // Neuron. 2010. V. 65. № 1. P. 66–79. https://doi.org/10.1016/j.neuron.2009.12.023
Scott D.A., Tabarean I., Tang Y. et al. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration // J. Neurosci. 2010. V. 30. № 24. P. 8083–8095. https://doi.org/10.1523/JNEUROSCI.1091-10.2010
Jowaed A., Schmitt I., Kaut O., Wullner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains // J. Neurosci. 2010. V. 30. № 18. P. 6355–6359. https://doi.org/10.1523/JNEUROSCI.6119-09.2010
Matsumoto L., Takuma H., Tamaoka A. et al. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease // PLoS One. 2010. V. 5. № 11. P. e15522. https://doi.org/10.1371/journal.pone.0015522
Ai S.X., Xu Q., Hu Y.C. et al. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease // J. Neurol. Sci. 2014. V. 337. № 1–2. P. 123–128. https://doi.org/10.1016/j.jns.2013.11.033
Tan Y.Y., Wu L., Zhao Z.B. et al. Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients // Parkinsonism Relat. Disord. 2014. V. 20. № 3. P. 308–313. https://doi.org/10.1016/j.parkreldis.2013.12.002
Pihlstrøm L., Berge V., Rengmark A., Toft M. Parkinson’s disease correlates with promoter methylation in the α-synuclein gene // Mov. Disord. 2015. V. 30. № 4. P. 577–580. https://doi.org/10.1002/mds.26073
Schmitt I., Kaut O., Khazneh H. et al. L-dopa increases α-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro // Mov. Disord. 2015. V. 30. № 13. P. 1794–1801. https://doi.org/10.1002/mds.26319
Kwok J.B., Teber E.T., Loy C. et al. Tau haplotypes regulate transcription and are associated with Parkinson’s disease // Ann. Neurol. 2004. V. 55. № 3. P. 329–334. https://doi.org/10.1002/ana.1082614991810
Vilarino-Güell C., Soto-Ortolaza A.I., Rajput A. et al. MAPT H1 haplotype is a risk factor for essential tremor and multiple system atrophy // Neurology. 2011. V. 76. № 7. P. 670–672. https://doi.org/10.1212/WNL.0b013e31820c30c1
Ezquerra M., Pastor P., Gaig C. et al. Different MAPT haplotypes are associated with Parkinson’s disease and progressive supranuclear palsy // Neurobiol. Aging. 2011. V. 32. № 3. 547. P. e11-6. https://doi.org/10.1016/j.neurobiolaging.2009.09.011
Caillet-Boudin M.L., Buée L., Sergeant N., Lefebvre B. Regulation of human MAPT gene expression // Mol. Neurodegener. 2015. V. 10: 28. P. 1–14. https://doi.org/10.1186/s13024-015-0025-8
Gilman S., Wenning G.K., Low P.A. et al. Second consensus statement on the diagnosis of multiple system atrophy // Neurology. 2008. V. 71. № 9. P. 670–676. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Coupland K.G., Mellick G.D., Silburn P.A. et al. DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E in vitro // Mov. Disord. 2014. V. 29. P. 1606–1614. https://doi.org/10.1002/mds.25784
Iwata A., Nagata K., Hatsuta H. et al. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation // Hum. Mol. Genet. 2014. V. 23. № 3. P. 648–656. https://doi.org/10.1093/hmg/ddt451
Hoffmann A., Sportelli V., Ziller M., Spengler D. Driver or passenger: Epigenomes in Alzheimer’s disease // Epigenomes. 2017. V. 1. № 1. 5. P. 1–18. https://doi.org/10.3390/epigenomes1010005
Li Y., Chen J.A., Sears R.L. et al. An epigenetic signature in peripheral blood associated with the haplotype on 17q21.31, a risk factor for neurodegenerative tauopathy // PLoS Genet. 2014. V. 10. № 3. P. e1004211.
Дополнительные материалы отсутствуют.