

Генетика, 2022, T. 58, № 9, стр. 1085-1093
Анализ мутаций гена ATL1 и клинических особенностей течения заболевания у пациентов с наследственной спастической параплегией
И. М. Хидиятова 1, 2, 3, *, Е. В. Сайфуллина 4, А. С. Карунас 1, 2, 3, А. Ф. Ахметгалеева 4, Р. Ф. Кутлубаева 4, Л. А. Смакова 1, С. Л. Лобов 1, А. В. Поляков 5, О. А. Щагина 5, В. А. Кадникова 5, О. П. Рыжкова 5, Р. В. Магжанов 4, Э. К. Хуснутдинова 1, 2, 3
1 Институт биохимии и генетики Уфимского федерального исследовательского центра
Российской академии наук
450054 Уфа, Россия
2 Башкирский государственный университет
450076 Уфа, Россия
3 Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
4 Башкирский государственный медицинский университет
450000 Уфа, Россия
5 Медико-генетический научный центр
115522 Москва, Россия
* E-mail: imkhid@mail.ru
Поступила в редакцию 01.02.2022
После доработки 15.03.2022
Принята к публикации 21.03.2022
- EDN: KSXEKY
- DOI: 10.31857/S0016675822090119
Аннотация
Наследственные спастические параплегии (НСП) – группа нейродегенеративных болезней с преимущественным поражением пирамидного тракта. Аутосомно-доминантная форма SPG3А, связанная с мутациями в гене ATL1, относится к наиболее распространенным в популяциях Европы формам НСП. В 63 неродственных семьях с НСП из Республики Башкортостан (РБ) проведен анализ гена ATL1. Идентифицировано два патогенных варианта: у одного пациента – дупликация всего 3-го экзона, у семи больных из трех неродственных семей – миссенс-мутация с.1246С>T (p.Arg416Cys). Частота распространения SPG3A среди неродственных пациентов в РБ составила 6.3%. В одной из обследованных семей установлено происхождение мутации с.1246С>T de novo. Клиническая характеристика заболевания в большинстве случаев соответствовала неосложненной НСП, протекающей в легкой форме. Выявлены внутрисемейные различия по клиническим проявлениям заболевания, в том числе у однояйцевых близнецов. Возраст манифестации у большинства обследованных пациентов составил от 10 до 50 лет.
Наследственные спастические параплегии (НСП) – генетически и клинически гетерогенная группа дегенеративных заболеваний нервной системы, обусловленных дистальным поражением длинных аксонов кортикоспинального тракта [1]. Основным клиническим проявлением НСП является спастичность мышц нижних конечностей. Спастические параличи могут сочетаться с различными дополнительными симптомами – атрофией зрительных нервов, глухотой, атаксией, полинейропатией, эпилепсией, нарушением когнитивных функций и др. В зависимости от того, является ли основной симптом единственным или сочетается с другими неврологическими или экстраневральными симптомами, выделяют неосложнeнные (“чистые”) или осложненные формы заболевания [2, 3]. Распространенность НСП в мире варьирует от 1.27 до 9.6 на 100 000 населения [1].
Значительная клиническая гетерогенность заболевания связана с разнообразными патогенетическими процессами, возникающими в нейронах: дефектами формирования мембранных органоидов, молекулярного транспорта, нарушением процессов миелинизиции, функций митохондрий, за которые ответственны мутации в различных генах. В настоящее время известно более 100 генов, связанных с НСП [http://www.neuromuscular.wustl.edu, октябрь 2021]. По современной номенклатуре, генные локусы и соответствующие формы НСП обозначают аббревиатурой SPG (от англ. Spastik Paraplegia Gene), с порядковыми номерами в хронологической последовательности [1]. Заболевание может наследоваться как по аутосомно-доминантному, так и по аутосомно-рецессивному и Х-сцепленному типам наследования. В популяциях Европы за 40–45% случаев аутосомно-доминантных НСП ответственны мутации в гене спастина (SPAST, SPG4) [4], за 10% случаев – в гене атластина (ATL1, SPG3A) [5]. Белковые продукты этих двух генов вовлечены во многие важные жизненные процессы в клетке и, в частности, они совместно координируют морфогенез эндоплазматического ретикулума и динамику микротрубочек, и нарушение этих процессов считается одним из главных предполагаемых механизмов аксональной дегенерации при НСП [6–11].
Генетическая форма SPG3A, связанная с мутациями в гене ATL1, зарегистрирована во многих популяциях мира и считается третьей по частоте среди аутосомно-доминантных НСП и наиболее распространенной причиной рано начинающейся НСП [5, 12–18]. В гене ATL1, локализованном в хромосомной области 14q22.1, на сегодняшний день зарегистрировано более 80 различных мутаций, 69 из которых – миссенс-мутации, гораздо реже встречаются небольшие делеции и инсерции, мутации сайта сплайсинга, а также делеции целых экзонов (HGMD professional 2022.4, http://www.hgmd.cf.ac.uk). Однако каких-либо четких клинических особенностей SPG3A до сих пор не выявлено, отмечается ее значительная клиническая гетерогенность, в том числе внутрисемейная. Единственным важным ориентиром пока считается ранний возраст манифестации заболевания (<10 лет), но он тоже не является абсолютным показателем, и даже в одной семье при одинаковой мутации в гене ATL1 могут наблюдаться как ранний, так и значительно более поздний дебют болезни [19]. Для некоторых типов мутаций гена ATL1, таких как делеции и инсерции, установление клинико-генетических корреляций пока невозможно в силу редкости таких случаев, и информация о них носит лишь описательный характер. Поэтому накопление данных о различных генетических вариантах НСП, в том числе об SPG3A, а также о их распространенности в различных популяциях не теряет своей актуальности. Детальные сведения о разнообразии генных мутаций и их функциональной роли в развитии болезни являются основой для разработки патогенетических методов лечения, а информация о распространенности типов генетически гетерогенных патологий, таких как НСП, о спектре и частоте мутаций в соответствующих генах позволяет разрабатывать наиболее эффективные, оптимальные для отдельных регионов, подходы ДНК-диагностики, в целом улучшая медико-генетическое консультирование в семьях пациентов.
В Республике Башкортостан, одном из многонациональных регионов России, эпидемиологическое и молекулярно-генетическое изучение наследственных спастических параплегий проводится уже в течение нескольких лет. Общая распространенность этой группы заболеваний в республике составляет 3.5 на 100 000 населения [20]. Ранее мы представляли сведения об отдельных случаях генетической формы SPG4, обусловленной мутациями в гене SPAST, вклад которой в общую структуру заболевания в РБ составил 33.3% [21–23]. В настоящей работе мы приводим результаты исследования гена ATL1 у пациентов с НСП из нашего региона.
МАТЕРИАЛЫ И МЕТОДЫ
Общая обследуемая выборка пациентов представлена 130 индивидами из 63 неродственных семей, что составляет около 70% всех зарегистрированных в Республике Башкортостан больных НСП. По этнической принадлежности в выборку вошли 27 татарских семей, 14 – русских, 5 – башкирских, по одной семье – чувашской, украинской, марийской, 8 – метисных семей, а также 6 семей с неустановленной этнической принадлежностью. В 39 семьях установлен аутосомно-доминантный тип наследования спастической параплегии, в двух – аутосомно-рецессивный, шесть пациентов имели спорадический характер болезни, а для 16 больных тип наследования точно установить не удалось. Выборка была сформирована на основе данных о больных, состоящих на диспансерном учете в Республиканском медико-генетическом центре (РМГЦ), и материалов ежегодных отчетов неврологической службы РБ за 2000–2018 гг., предоставляемых ЦРБ и ЛПУ г. Уфы в распоряжение Медико-информационного аналитического центра МЗ РБ. Больные и их ближайшие родственники были осмотрены сотрудниками кафедры неврологии с курсами нейрохирургии и медицинской генетики Башкирского государственного медицинского университета. Контрольная выборка состояла из здоровых жителей РБ различной этнической принадлежности: русской (50 чел.), татарской (50 чел.), башкирской (50 чел.). ДНК была выделена из крови методом фенольно-хлороформной экстракции [24].
Анализ гена ATL1 был проведен у всех 63 неродственных пациентов, независимо от того, были ли у них до этого идентифицированы мутации в гене спастина. Поскольку выборка пациентов формировалась и одновременно исследовалась на протяжении нескольких лет, анализ гена ATL1 у разных пациентов был проведен разными методами: у первых 57 неродственных больных поиск мутаций был проведен методом конформационного полиморфизма однонитевой ДНК (SSCP-анализ) и с целью выявления протяженных делеций или инсерций – методом мультиплексной лигазозависимой амплификации (MLPA-анализ); у шести “новых” пациентов с аутосомно-доминантной НСП с неустановленной генетической причиной заболевания было проведено массовое параллельное секвенирование с использованием таргетной панели, включающее анализ кодирующих последовательностей 63 генов, ответственных за различные типы НСП.
Методом SSCP-анализа исследовались все 14 экзонов с прилегающими интронными областями гена ATL1. Амплификацию соответствующих фрагментов ДНК проводили методом ПЦР на амплификаторах T100 Thermal Cycler (Bio Rad, США) и Thermal Cycler 2720 (Applied Biosystems, США), с использованием олигонуклеотидных праймеров, представленных в работе [15].
MLPA-анализ гена ATL1 проводили с использованием набора реактивов MLPA probemix P165, MRC-Holland. Реакции MLPA проводили в соответствии с инструкциями производителя. Электрофорез продуктов ПЦР выполняли с использованием генетического анализатора ABI 3130 (Applied Biosystems). Данные MLPA анализировали в программе Coffalyser.Net. Программа рассчитывает относительную высоту пиков образцов и контролей (соотношение пиков). На гетерозиготную делецию экзона указывает отношение относительной высоты пика между образцом и контролем ниже 0.7, тогда как отношение относительной высоты пика выше 1.3 указывает на дупликацию или умножение экзона.
Таргетное секвенирование экзома проведено на секвенаторе нового поколения Ion S5. Для пробоподготовки использована технология ультрамультиплексной ПЦР, сопряженная с последующим секвенированием (AmpliSeq™). Анализ проведен с использованием кастомной панели “Спастические параплегии”, включающей кодирующие последовательности генов: GJC2, AP4B1, AMPD2, IBA57, ALDH18A1, ZFYVE27, NT5C2, ENTPD1, MTPAP, CAPN1, BSCL2, KLC2, KIF5A, C12orf65, MARS, VAMP1, B4GALNT1, SPG20, SACS, ATL1, ZFYVE26, DDHD1, TECPR2, AP4S1, NIPA1, SPG11, SPG21, AP4E1, USP8, SPG7, FA2H, ARL6IP1, KIF1C, AFG3L2, RTN2, PNPLA6, C19orf12, CPT1C, MAG, HSPD1, KIF1A, REEP1, PGAP1, MARS2, SPAST, SLC33A1, TFG, WDR48, CYP2U1, ARSI, ZFR, REEP2, AP5Z1, AP4M1, CYP7B1, KIAA0196, ERLIN2, VPS37A, DDHD2, GBA2, L1CAM, PLP1, SLC16A2. По данным AmpliSeq™ Сoverage Analysis, среднее покрытие панели “Спастические параплегии” составляет 363.1, Uniformity 92.02%. Обработка данных секвенирования проведена с использованием стандартного автоматизированного алгоритма, предлагаемого TermoFisher Scientific (Torrent Suite™), а также программного обеспечения Gene-Talk.
Для оценки популяционных частот выявленных вариантов использованы выборки проектов “1000 геномов”, ESP6500 и Genome Aggregation Database (gnomAD). Для оценки клинической релевантности выявленных вариантов использованы база данных OMIM, база данных по патогенным вариантам HGMD® Professional, версия 2020.4.
Для подтверждения выявленной в результате NGS-секвенирования мутации с.1246C>T в гене ATL1 у пробанда проведено секвенирование 12-го экзона гена методом Сэнгера на автоматическом секвенаторе ABI PRISM 3130 XL (Applied Biosystems). Скрининг на наличие/отсутствие данной мутации у шести членов семьи пробанда (больной сын и пять здоровых родственников), а также у пациентов с НСП из 62 неродственных семей и в популяционных выборках здоровых индивидов проведен методом ПДРФ-анализа с использованием эндонуклеазы MwoI.
РЕЗУЛЬТАТЫ
В результате SSCP-анализа 14 экзонов гена ATL1, проведенного у 57 пациентов с НСП из РБ, не было выявлено вариантов, которые могли бы расцениваться как патогенные или вероятно патогенные.
В результате MLPA-анализа, проведенного у этих же больных, в одном случае у пациентки Г. (33.3) из русской семьи была идентифицирована дупликация третьего экзона (dup 3 ex) гена ATL1 в гетерозиготном состоянии (рис. 1).
Рис. 1.
Результаты MLPA-анализа после обработки в Cofalyser.net: идентификация дупликации третьего экзона гена ATL1 (зонд ATL1-3-168nt).

Заболевание в семье пациентки Г. наследуется с доминантным паттерном, однако по объективным причинам у других больных членов семьи ДНК-анализ провести не удалось. Первыми симптомами НСП у пробанда были слабость в ногах, изменение походки. В клинической картине: походка изменена по типу спастико-атактической. Сила мышц ног снижена (проксимально), мышечный тонус повышен, при этом снижена глубокая чувствительность в ногах. Кроме данных симптомов, у пациентки выявляется дисфункция тазовых органов. Все симптомы соответствуют неосложненной форме заболевания. Возраст начала болезни – около 40 лет.
В результате таргетного секвенирования панели генов у пациента К. (51.0) татарской этнической принадлежности с аутосомно-доминантной НСП в 12-м экзоне гена ATL1 был идентифицирован в гетерозиготном состоянии миссенс-вариант с.1246С>T (p.Arg416Cys), подтвержденный методом секвенирования по Сэнгеру (рис. 2).
Рис. 2.
Секвенирование 12-го экзона гена ATL1 у пациента с НСП: нуклеотидная замена с.1246С>T в гетерозиготном состоянии.
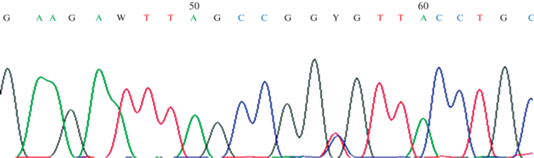
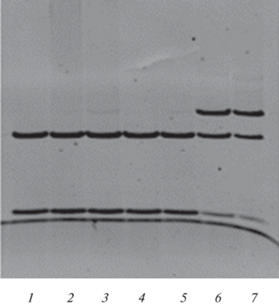
Данный нуклеотидный вариант является известной патогенной мутацией, он был описан ранее у пациентов с НСП [25–27]. Эта мутация может быть идентифицирована методом ПДРФ-анализа с использованием эндонуклеаз NgoMIV, Cac8I, NaeI и MwoI, для которых исчезает существующий в норме сайт рестрикции. Этим методом, с использованием рестриктазы MwoI (рис. 3), мы провели поиск мутации с.1246С>T у других членов семьи пробанда, у других неродственных пациентов общей обследуемой выборки, а также в контрольных выборках здоровых индивидов (150 чел.).
Рис. 3.
Образец идентификации мутации с.1246С>T (p.Arg416Cys) в гене ATL1 методом ПДРФ-анализа с использованием эндонуклеазы MwoI (7%-ный ПААГ): 1–5 – образцы ДНК без мутации; 6, 7 – образцы ДНК с мутацией в гетерозиготном состоянии.
В семье пациента К. (51.0) нуклеотидная замена с.1246С>T в гене ATL1 была выявлена также у его больного сына и не обнаружена у здоровых членов семьи – брата, сестры, дочери, и ни у одного из родителей. На основании отсутствия мутации у родителей было сделано предположение, что у пробанда мутация возникла de novo. Проведенный микросателлитный анализ подтвердил кровное родство пробанда с обоими родителями, что подтвердило данное предположение.
В результате скрининга на наличие мутации с.1246С>T у других пациентов из обследуемой выборки она была выявлена еще в двух неродственных семьях – у пациента Л. (16.0) русской этнической принадлежности, у его матери и у его сына, и у двух сестер-близнецов Ч. (41.01 и 41.02) метисного происхождения (мордва/татарка). В контрольных популяционных выборках здоровых индивидов данная мутация не обнаружена.
У всех доступных для исследования пациентов из трех семей с мутацией с.1246С>T проведен анализ клинической картины НСП.
Пробанд 51.0, 1967 г. рождения, с 10–12-летнего возраста отмечал затруднения при быстрой ходьбе и беге, связывал эти жалобы с особенностью строения стоп (“плоские”). В конце второго–начале третьего десятилетия жизни пациента окружающие начали замечать изменение его походки: “шаркает ногами”. Позднее, ближе к 40 годам, сам пациент стал отчетливо ощущать скованность в мышцах ног, приводящую к изменению походки и утомляемости при длительной ходьбе. При объективном осмотре больного выявлено повышение мышечного тонуса, преимущественно в мышцах-сгибателях голеней и стоп по спастическому типу до трех баллов по шкале Ашворта, снижение мышечной силы до четырех баллов в проксимальных отделах ног. “Полые” стопы. Сухожильные рефлексы в ногах повышены, отмечаются клонусы обеих стоп и патологические стопные знаки сгибательного и разгибательного типов. Ходит самостоятельно, походка изменена по типу “лыжника”. У сына пробанда, 1990 г. рождения, клинически осмотренного в возрасте 30 лет, отмечаются подобное изменение походки, повышение мышечного тонуса в ногах до одного балла, а также сухожильных рефлексов с ног, клонусы обеих стоп и патологические стопные знаки.
В семье Л. под нашим наблюдением находились пробанд (16.0), его мать и его сын. Первые признаки болезни пробанд отметил в возрасте 20 лет, когда начал замечать изменение походки из-за ощущения скованности в ногах. При осмотре пациента в возрасте 30 лет клиническая симптоматика соответствовала неосложненной спастической параплегии: нижний спастический парапарез, гиперрефлексия сухожильных рефлексов, положительные стопные и кистевые патологические рефлексы, легкое снижение вибрационной чувствительности в ногах, дисфункция тазовых органов по типу задержки мочеиспускания. У матери пробанда активных жалоб на момент осмотра (в возрасте 56 лет) не было, ее походка была не изменена, но сухожильные рефлексы с ног были повышены и отмечались разгибательные стопные патологические рефлексы. Сын пробанда был осмотрен в возрасте 16 лет, активных жалоб на здоровье не предъявлял, походка была не изменена, мышечный тонус в ногах был удовлетворительным, но отмечалась сухожильная гиперрефлексия с ног. Таким образом, заболевание в семье представлено неосложненной формой с вариабельной экспрессивностью.
У однояйцевых сестер Ч. (41.01 и 41.02) отмечалась разница как по возрасту манифестации болезни, так и по спектру клинических симптомов. К сожалению, провести осмотр родителей, а также других сибсов и детей сестер не удалось по объективным причинам. У первой сестры изменение походки появилось в возрасте 43–44 лет, клиническая картина в возрасте 46 лет соответствовала неосложненной форме заболевания; у второй сестры жалобы на изменение походки отмечены с возраста 49–50 лет, клиническая симптоматика в возрасте 53 лет включала помимо нижнего спастического парапареза легкое снижение вибрационной чувствительности в ногах и снижение когнитивных функций умеренной степени. Электрофизиологические параметры (скорости распространения возбуждения и амплитуды М-ответов по периферическим нервам рук и ног) были в пределах нормальных величин; нейровизуализационные параметры спинного мозга – в норме, а по данным МРТ головного мозга – признаки микроангиопатии и начальные проявления атрофического процесса. Для уточнения связи между снижением когнитивных функций и основным заболеванием требуется продолжение обследования как самой пациентки, так и ее однояйцевой сестры.
ОБСУЖДЕНИЕ
В обследуемой нами выборке пациентов с НСП из Республики Башкортостан в четырех из 63 неродственных семей в гене ATL1 были идентифицированы два различных изменения нуклеотидной последовательности: в одной семье – ранее неописанная дупликация 3-го экзона гена, в трех семьях – известная миссенс-мутация с.1246С>T (p.Arg416Cys). Во всех этих семьях установлен аутосомно-доминантный тип наследования НСП. У пациента с дупликацией 3-го экзона наблюдалась неосложненная форма заболевания, сопровождающаяся дисфункцией тазовых органов, проявившаяся в возрасте 40 лет. У семи обследованных пациентов из трех семей с мутацией p.Arg416Cys возраст манифестации болезни варьировал от 10 до 50 лет, у большинства из них клиническая картина соответствовала неосложненной форме заболевания, в одном случае – сочетающаяся с дисфункцией тазовых органов; у одной пациентки основные клинические симптомы НСП осложнены когнитивным расстройством умеренной степени. Тяжесть заболевания у большинства из наших пациентов НСП легкая, отмечены и практически бессимптомные случаи (2/7 – 28.6%).
Как уже было сказано, при SPG3А наиболее распространенными мутациями в гене ATL1 являются миссенс-варианты, тогда как делеции или инсерции встречаются гораздо реже, и в единственном случае описана делеция экзона: Sulek с соавт. [28] методом MLPA-анализа провели скрининг 93 пациентов на наличие протяженных делеций/дупликаций в генах SPAST и ATL1, выявив 11 различных делеций и одну дупликацию в гене спастина и делецию 4-го экзона в гене атластина. О полной делеции одного аллеля гена ATL1 в гетерозиготном состоянии, сочетающейся с делецией экзона гена SPAST, еще ранее сообщили Beetz et al. [29], в то же время предположив, что у обследованного ими пациента заболевание было обусловлено мутацией гена SPAST, а не делецией целого гена ATL1. Кодируемый геном ATL1 трансмембранный белок атластин-1 принадлежит подсемейству динаминов больших ГТФаз, имеет N-концевой ГТФ-связывающий домен и два близкорасположенных гидрофобных сегмента на С-конце, которые формируют трансмембранный домен [8, 9]. Третий экзон гена ATL1 соответствует ГТФазному домену белка. В этой области находится каталитически активная глутаминовая кислота в 117-м положении, которая участвует в гидролизе ГТФ [30]. На сегодняшний день аутосомно-доминантных случаев заболевания, связанных с патогенными вариантами в третьем экзоне гена, неизвестно, и описаны только две мутации – p.R118Q и p.R217*, идентифицированные в гомозиготном состоянии в семьях с аутосомно-рецессивной формой НСП [31, 32]. Обе эти мутации расположены в консервативном нуклеотид-связывающем сайте ГТФазного домена и предполагается, что они приводят к потере функции гена таким образом, что это не мешает аллелю дикого типа. У всех пациентов из семей c данными мутациями зарегистрирован ранний возраст манифестации – 1-й год жизни, неосложненная форма НСП, с развитием в третьей декаде жизни дисфункции тазовых органов, в частности недержания мочи [31, 32]. Большинство клинических признаков, описанных у больных членов этих семей, совпадает с таковыми, выявленными у нашей пациентки с дупликацией 3-го экзона, кроме гораздо более позднего у нее возраста манифестации НСП – около 40 лет. Исходя из сведений о функциональной значимости данной области гена, можно предположить, что протяженная дупликация третьего экзона гена ATL1, выявленная нами в гетерозиготном состоянии у пациента с НСП, может оказывать значительный негативный эффект на ГТФазную функцию атласина, когда компенсация функции за счет аллеля дикого типа является неполной. Такой эффект мутации может быть связан с разными механизмами. В случае, если дупликация экзона представляет собой вставку нуклеотидной последовательности по количеству нуклеотидов некратному трем, то очевиден сдвиг рамки считывания, нарушающий всю кодирующую последовательность гена, как правило приводящий к образованию преждевременного стоп-кодона и как следствие – к нонсенс-опосредованному разрушению белка. Если же количество нуклеотидов вставки кратно трем, то предположительно негативный эффект может быть связан со значительным размером ее кодирующей части, причем с большой вероятностью того, что в этой части будут другие аминокислоты, так как начало экзона не всегда совпадает с триплетом. Однако эти заключения являются предположительными и требуют дальнейших исследований, поскольку мутация была выявлена методом MLPA-анализа, не позволяющим установить точные границы инсерции.
Нуклеотидная замена с.1246С>T (p.Arg416Cys) в гене ATL1 ранее была описана Orlacchio et al. [25], Magariello et al. [26] и Luo et al. [27] как патогенная мутация, обусловливающая НСП формы SPG3A с аутосомно-доминантным типом наследования. Клиническая картина НСП у пациентов, представленных в этих работах, несколько различается. Так, Orlacchio et al. [25] идентифицировали эту мутацию у 10 пациентов с АД НСП из одной южноафриканской семьи: у них у всех отмечался поздний возраст манифестации заболевания (от 39 до 51 года), а также у всех – умственная отсталость (IQ от 32 до 67).
Magariello et al. [26] такую же мутацию выявили у пациентки с НСП, у которой заболевание было впервые диагностировано в возрасте 40 лет, однако его первые признаки в виде трудностей при беге, судорог нижних конечностей и поясничных болей сама пациентка отмечала с более раннего возраста – после полового созревания. Заболевание у данной пациентки соответствовало неосложненной НСП, в частности когнитивных нарушений у нее выявлено не было. По данным исследования Luo et al. [27], в одной китайской семье с НСП, обусловленной мутацией p.Arg416Cys, у большинства больных членов семьи заболевание в неосложненной форме развилось до 10 лет, но у некоторых – в более позднем возрасте. Обобщая приведенные выше сведения, можно заключить, что НСП, обусловленная мутацией с.1246С>T в 12-м экзоне гена ATL1, может проявляться как в раннем (до 10 лет), так и гораздо в более позднем возрасте, часто – после 40 лет. В большинстве случаев НСП соответствует неосложненному типу, в рамках которого иногда могут присутствовать снижение вибрационной чувствительности в ногах или признаки дисфункции тазовых органов в виде затруднения мочеиспускания или недержания мочи; реже у носителей данной мутации спастическая параплегия может быть осложнена когнитивными нарушениями. По клиническим проявлениям может наблюдаться как межсемейная, так и внутрисемейная вариабельность, даже в случае однояйцевых близнецов, что может свидетельствовать о роли эпигенетических факторов в развитии и течении болезни. В одной из наших семей установлено происхождение мутации с.1246С>T de novo, что подтверждает существующее предположение о наличии “горячей точки” в 12-м экзоне гена. Также наряду с 12-м экзоном “горячими” считаются экзоны 4, 7 и 8 гена ATL1, и большинство (87.32%) известных мутаций в гене приходится на эти экзоны [19].
Zhao и Liu [19] провели обобщенный анализ гено-фенотипической корреляции по сведениям о пациентах с SPG3A из 142 семей, представленным в 51 публикации. Авторы показали, что у 84.79% пациентов с мутациями в гене ATL1 наблюдается ранняя форма SPG3A, с возрастом манифестации <10 лет, и у 15.21% – более поздняя (>10 лет) форма заболевания. Это обстоятельство, подтверждаемое и нашими данными, подчеркивает необходимость анализа гена ATL1 не только при ранних формах НСП. Кроме того, было показано, что по возрасту начала болезни даже при одной и той же мутации может наблюдаться как межсемейная, так и внутрисемейная неоднородность (в частности, при мутациях p.A161P и p.R495W [33, 34]). Сравнение клинических данных, проведенное в этом же исследовании, не выявило какой-либо корреляции генотип–фенотип при различных мутациях в гене ATL1 [19]. Из 440 пациентов, для которых была доступна информация в исследовании этих авторов, у 378 больных (85.90%) была установлена “чистая” форма НСП и у 62 (14.10%) – осложненная форма болезни. Осложняющие симптомы у пациентов со спастической параплегией SPG3A включали судороги, атрофию зрительного нерва, сенсорные нарушения, умственную отсталость, атаксию, дистальную атрофию и периферическую аксональную невропатию. При этом авторы обнаружили, что дистальная атрофия является наиболее частым симптомом у пациентов с осложненными формами SPG3A. Включенные в клиническую картину неосложненной формы болезни снижение вибрационной чувствительности в ногах и дисфункция тазовых органов, выявленные в обследованной нами семье Л., при SPG3A встречаются гораздо реже, чем при других формах АД НСП [19].
Тяжесть заболевания у большинства пациентов с SPG3A отмечается как легкая, но степень спастичности увеличивается с продолжительностью заболевания. Пенетрантность заболевания составляет 80–90%, но в ряде семейных случаев у гетерозиготных носителей мутации в гене ATL1 в преклонном возрасте неврологический статус остается в норме, что свидетельствует против возраст-зависимой пенетрантности [15]. Самый низкий процент пенетрантности (30%) был описан в семье с мутацией в соседнем, 415-м, кодоне гена ATL1: c.1243C>T (p.Arg415Trp) [35].
В целом для мутаций в гене ATL1 пока сложно провести четкие гено-фенотипические корреляции, но накопление данных по конкретным патогенным вариантам может способствовать пониманию таких взаимосвязей. В частности, полученные нами данные свидетельствуют о более позднем возрасте манифестации НСП и ее относительно легком течении при выявленных у наших пациентов мутациях в 3-м и 12-м экзонах, подтверждают сведения о неполной пенетрантности мутации с.1246С>T и о существовании “горячей” точки в 12-м экзоне. Проведенное нами популяционное исследование также демонстрирует неоднородность распространения отдельных генетических форм НСП в различных регионах. В общей выборке из 63 пробандов с НСП, представителей, примерно, 70% всех зарегистрированных в Республике Башкортостан неродственных семей пациентов, вклад генетической формы SPG3 в структуру заболевания составил 6.3%, частота мутации с.1246С>T (p.Arg416Cys) – 4.7%. Эти данные дополняют общую картину по спектру и частоте патогенных вариантов, приводящих к развитию НСП у пациентов обследуемого региона, и способствуют повышению эффективности генетического консультирования в семьях пациентов.
Работа выполнена в рамках Государственного задания Минобрнауки РФ (№ АААА-А16-116020350032-1), при частичной финансовой поддержке Санкт-Петербургского государственного университета (проект № 93025749), гранта РФФИ (№ 17-44-020951) и Мегагранта Правительства Российской Федерации (соглашение № 075-15-2021-595). Для исследования использовано оборудование ЦКП “Биомика” (Отделение биохимических методов исследований и нанобиотехнологии РЦКП “Агидель”) и УНУ “КОДИНК”. Образцы ДНК для исследования взяты из “Коллекции биологических материалов человека” ИБГ УФИЦ РАН, поддержанной Программой биоресурсных коллекций ФАНО России (соглашение № 007-030164/2).
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики. Исследования были одобрены биоэтическим комитетом ИБГ УФИЦ РАН.
От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Fink J.K. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms // Acta Neuropathologica. 2013. V. 126. № 3. P. 307–328. https://doi.org/10.1007/s00401-013-1115-8
Hazan J., Fonknechten N., Mavel D. et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia // Nat. Genet. 1999. V. 23. № 3. P. 296–303. https://doi.org/10.1038/15472
Finsterer J., Löscher W., Quasthoff S. et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance // J. Neurological Sci. 2012. V. 318. № 1. P. 1–18. https://doi.org/10.1016/j.jns.2012.03.025
Fonknechten N., Mavel D., Byrne B. et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia // Human Mol. Genet. 2000. V. 9. № 4. P. 637–644. https://doi.org/10.1093/hmg/9.4.637
Namekawa M., Nelson I., Ribai P. et al. A founder effect and mutational hot spots may contribute to the most frequent mutations in the SPG3A gene // Neurogenetics. 2006. V. 7. № 2. P. 131–132. https://doi.org/10.1007/s10048-006-0028-2
Zhu P.P., Soderblom C., Tao-Cheng J.H. et al. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development // Human Mol. Genet. 2006. V. 15. № 8. P. 1343–1353. https://doi.org/10.1093/hmg/ddl054
Namekawa M., Muriel M.P., Janer A. et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis // Mol. and Cellular Neuroscience. 2007. V. 35. № 1. P. 1–13. https://doi.org/10.1016/j.mcn.2007.01.012
Rismanchi N., Soderblom C., Stadler J. et al. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis // Human Mol. Genet. 2008. V. 17. № 11. P. 1591–1604. https://doi.org/10.1093/hmg/ddn046
Hu J., Shibata Y., Zhu P.P. et al. A class of dynamin-like GTPases involved in the generation of the tubular ER network // Cell. 2009. V. 138. № 3. P. 549–561. https://doi.org/10.1016/j.cell.2009.05.025
Orso G., Pendin D., Liu S. et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin // Nature. 2009. V. 460. № 7258. P. 978–983. https://doi.org/10.1038/nature08280
Park S.H., Zhu P.-P., Parker R.L., Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network // The J. Clin. Investigation. 2010. V. 120. № 4. P. 1097–1110. https://doi.org/10.1172/JCI40979
Zhao X., Alvarado D., Rainier S. et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia // Nat. Genet. 2001. V. 29. № 3. P. 326–331. https://doi.org/10.1038/ng758
Tessa A., Casali C., Damiano M. et al. SPG3A an additional family carrying a new atlastin mutation // Neurology. 2002. V. 59. № 12. https://doi.org/10.1212/01.wnl.0000036902.21438.98
Abel A., Fonknechten N., Hofer A. et al. Early onset autosomal dominant spastic paraplegia caused by novel mutations in SPG3A // Neurogenetics. 2004. V. 5. № 4. P. 239–243. https://doi.org/10.1007/s10048-004-0191-2
Dürr A., Camuzat A., Colin E. et al. Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia // Arch. Neurol. 2004. V. 61. P. 1867–1872. https://doi.org/10.1001/archneur.61.12.1867
Hedera P., Fenichel G.M., Blair M., Haines J.L. Novel mutation in the SPG3A gene in an African American family with an early onset of hereditary spastic paraplegia // Arch. Neurol. 2004. V. 61. № 10. P. 1600–1603. https://doi.org/10.1001/archneur.61.10.1600
Sauter S.M., Engel W., Neumann L.M. et al. Novel mutations in the Atlastin gene (SPG3A) in families with autosomal dominant hereditary spastic paraplegia and evidence for late onset forms of HSP linked to the SPG3A locus // Hum. Mutat. 2004. V. 23. № 1. P. 98. https://doi.org/10.1002/humu.9205
Erfanian Omidvar M., Torkamandi S., Rezaei S. et al. Genotype–phenotype associations in hereditary spastic paraplegia: A systematic review and meta-analysis on 13,570 patients // J. Neurol. 2021. V. 268. P. 2065–2082. https://doi.org/10.1007/s00415-019-09633-1
Zhao G.-H., Liu X.-M. Clinical features and genotype–phenotype correlation analysis in patients with ATL1 mutations: a literature reanalysis // Transl. Neurodegen. 2017. V. 6. № 1. P. 9. https://doi.org/10.1186/s40035-017-0079-3
Магжанов Р.В., Сайфуллина Е.В., Идрисова Р.Ф. и др. Эпидемиологическая характеристика наследственных спастических параплегий в Республике Башкортостан // Мед. генетика. 2013. № 7. С. 12–16.
Ахметгалеева А.Ф., Хидиятова И.М., Сайфуллина Е.В. и др. Две новые мутации в гене SPG4 у пациентов с аутосомно-доминантной спастической параплегией из Республики Башкортостан // Генетика. 2016. Т. 52. № 6. С. 691–696.
Ахметгалеева А.Ф., Хидиятова И.М., Сайфуллина Е.В. и др. Клинический случай спорадической спастической параплегии при новой мутации в гене SPAST // Мед. генетика. 2016. № 7. С. 11–13.
Хидиятова И.М., Ахметгалеева А.Ф., Сайфуллина Е.В. и др. Мажорная мутация в гене SPAST у пациентов с аутосомно-доминантной спастической параплегией из Республики Башкортостан // Генетика. 2019. Т. 55. № 2. С. 229–233.
Mathew C.G.P. The isolation of high molecular weight eukaryotic DNA // Nucl. Acids. Res. 1984. P. 31–34.
Orlacchio A., Montieri P., Babalini C. et al. Late-onset hereditary spastic paraplegia with thin corpus callosum caused by a new SPG3A mutation // J. Neurol. 2011. V. 258. № 7. P. 1361–1363. https://doi.org/10.1007/s00415-011-5934-z
Magariello A., Tortorella C., Citrigno L. et al. The p.Arg416Cys mutation in SPG3a gene associated with a pure form of spastic paraplegia // Muscle Nerve. 2012. V. 45. № 6. P. 919–920. https://doi.org/10.1002/mus.23360
Luo Y., Chen C., Zhan Z. et al. Mutation and clinical characteristics of autosomal-dominant hereditary spastic paraplegias in China // Neurodegen. Dis. 2014. V. 14. № 4. P. 176–183. https://doi.org/10.1159/000365513
Sulek A., Elert E., Rajkiewic M. et al. Screening for the hereditary spastic paraplaegias SPG4 and SPG3A with the multiplex ligation-dependent probe amplification technique in a large population of affected individuals // Neurol. Sci. 2012. V. 34. № 2. P. 239–242. https://doi.org/10.1007/s10072-011-0899-3
Beetz C., Nygren A.O.H., Deufel T., Reid E. An SPG3A whole gene deletion neither co-segregates with disease nor modifies phenotype in a hereditary spastic paraplegia family with a pathogenic SPG4 deletion // Neurogenetics. 2007. V. 8. P. 317–318. https://doi.org/10.1007/s10048-007-0099-8
Bian X., Klemm R.W., Liu T.Y. et al. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes // Proc. Natl Acad. Sci. USA. 2011. V. 108. № 10. P. 3976–3981. https://doi.org/10.1073/pnas.1101643108
Khan T.N., Klar J., Tariq M. et al. Evidence for autosomal recessive inheritance in SPG3A caused by homozygosity for a novel ATL1 missense mutation // Eur. J. Hum. Genet. 2014. V. 22. P. 1180–1184. https://doi.org/10.1038/ejhg.2014.5
Willkomm L., Heredia R., Hoffmann K. et al. Homozygous mutation in Atlastin GTPase 1 causes recessive hereditary spastic paraplegia // J. Hum. Genet. 2016. V. 61. № 6. P. 571–573. https://doi.org/10.1038/jhg.2016.6
Scarano V., Mancini P., Criscuolo C. et al. The R495W mutation in SPG3A causes spastic paraplegia associated with axonal neuropathy // J. Neurol. 2005. V. 252. P. 901–903. https://doi.org/10.1007/s00415-005-0768-1
Ivanova N., Claeys K.G., Deconinck T. et al. Hereditary spastic paraplegia 3A associated with axonal neuropathy // Arch. Neurol. 2007. V. 64. P. 706–713. https://doi.org/10.1001/archneur.64.5.706
D’Amico A., Tessa A., Sabino A. et al. Incomplete penetrance in an SPG3A-linked family with a new mutation in the atlastin gene // Neurology. 2004. V. 62. P. 2138–2139. https://doi.org/10.1212/01.wnl.0000127698.88895.85
Дополнительные материалы отсутствуют.