

Генетика, 2023, T. 59, № 12, стр. 1393-1406
Популяционно-генетическая структура белого медведя (Ursus maritimus) в морях российской Арктики
П. А. Сорокин 1, *, Е. Ю. Звычайная 1, Е. А. Иванов 1, И. А. Мизин 2, И. Н. Мордвинцев 1, Н. Г. Платонов 1, А. И. Исаченко 3, Р. Е. Лазарева 3, В. В. Рожнов 1
1 Институт проблем экологии и эволюции им. А.Н. Северцова Российской академии наук
119071 Москва, Россия
2 Национальный парк “Русская Арктика”
163051 Архангельск, Россия
3 ООО “Арктический Научный Центр”
119333 Москва, Россия
* E-mail: sorokin-p@yandex.ru
Поступила в редакцию 30.05.2023
После доработки 16.07.2023
Принята к публикации 18.07.2023
- EDN: VDKHCW
- DOI: 10.31857/S0016675823120123
Аннотация
Рассмотрена популяционно-генетическая структура белого медведя (Ursus maritimus) на модельных участках в морях российской Арктики по материалам, собранным в период 2010–2021 гг. Получены данные по полиморфизму 17 микросателлитных локусов ядерной ДНК и фрагмента D-петли мтДНК длиной 610 пн для 93 животных. Для исследованной выборки взрослых белых медведей обнаружено высокое генетическое разнообразие ядерной ДНК и низкое значение нуклеотидной изменчивости π по митохондриальной ДНК. По всем генетическим маркерам обнаружена дифференциация медведей из южной части Баренцева моря от животных севера Баренцева и Карского морей. Данные группировки различаются по распределению митохондриального маркера (θst = 0.270) и слабо дифференцируются по ядерным локусам (Rst = 0.018).
Белый медведь (Ursus maritimus Phipps, 1774) – эволюционно относительно молодой вид, имеющий циркумполярный ареал и адаптировавшийся к жизни и охоте на льдах [1, 2]. Международные природоохранные организации, исходя из соображений удобства природоохранного менеджмента, рассматривают всех белых медведей как единую популяцию, разделенную на 19 субпопуляций [3].
Многочисленные исследования внутривидовой структуры белого медведя с использованием различных генетических методов (секвенирования фрагментов мтДНК, фрагментного анализа микросателлитных локусов, исследования однонуклеотидного полиморфизма SNP) и перемещений животных с помощью радиотелеметрии и спутниковых передатчиков показали различную степень дифференциации популяций – от слабовыраженных до статистически значимых различий, подтверждающих их пространственную обособленность. Так, анализ перемещений белых медведей и их генетической изменчивости выявил резкий контраст между их группировками с плохо выраженной генетической структурой, окружающими Полярный бассейн, и четырьмя генетическими изолятами в канадской Арктике, соответствующими отдельным популяциям медведей. Предполагается, что такие различия обусловлены сезонным распределением морского льда и тюленей [4]. Группировки медведей Гудзонова залива и Канадского Арктического архипелага генетически хорошо различаются, при этом особый интерес представляет небольшая генетически изолированная группировка Норвежского залива, ограниченного толстым льдом, сушей и полыньями [4, 5]. А наименее генетически различающимися оказались белые медведи Чукотского моря и моря Бофорта. Данные радиотелеметрии показали, что хотя они выделены в отдельные субпопуляции, участки обитания животных в них перекрываются, а это предполагает существование здесь единой популяции белых медведей [6].
В последнее время генетическая структура глобальной популяции белых медведей пересматривается. Одни авторы [7] выделяют три–четыре основных генетических кластера: Канадский Арктический архипелаг, Южная Канада и Полярный бассейн (последний подразделяется на восточный и западный субкластеры). При этом в связи с происходящими изменениями климата предполагается существование направленного потока генов, идущего преимущественно через самцов, из генетического кластера южной Канады и восточного субкластера Полярного бассейна в генетический кластер Канадского Арктического архипелага, который рассматривается в качестве межледникового рефугиума. Другие авторы [8] выделяют шесть генетических кластеров белых медведей: Гудзонова залива, западного и восточного участков Канадского Арктического архипелага, западного и восточного Полярного бассейнов и Норвежского залива. Хотя на численность, распространение и структуру популяции белых медведей негативно влияет быстрая потеря арктического морского льда, эти генетические данные, в отличие от предыдущей работы, не подтверждают существования сильного направленного потока генов в ответ на происходящее изменение климата.
В акваториях Карского и Баренцева морей белые медведи встречаются практически повсеместно, за исключением юго-западной части Баренцева моря, которое также рассматривается в качестве межледникового рефугиума. На основе данных телеметрии в этом регионе выделяют несколько обособленных группировок этого вида: две на севере Баренцева моря и две (северную и южную) в Карском море. Имеющиеся данные о перемещениях белых медведей в этом регионе свидетельствуют об относительной обособленности белых медведей, обитающих в районе острова Южный архипелага Новая Земля [9]. Подавляющее большинство данных о перемещениях получено для взрослых самок, которые привержены достаточно четко ограниченным местообитаниям и местам обустройства родовых берлог [10, 11]. При такой выраженной и стабильной пространственной структуре группировок белого медведя остаются неизученными механизмы, которые обеспечивают перемешивание и генетическую однородность мировой популяции. Возможно, она поддерживается за счет расселения молодых особей или миграции самцов, о чем известно значительно меньше, чем о перемещениях самок. Отдельные работы, выполненные на небольшом количестве животных, показывают, что самцы могут перемещаться более активно, оставаясь при этом в пределах территории, характерной для локальной группировки самок [10], или даже используя меньшую территорию [12]. Однако данные о перемещении самцов обычно ограничены несколькими месяцами с момента мечения животного [10, 13]. Имеющиеся данные о генетической структуре и о перемещениях белых медведей карско-баренцевоморского региона были получены в основном на медведях из северной части Баренцева моря. Группировка медведей, обитающая в южной части Баренцева моря, ранее не исследовалась.
Цель данной работы состояла в оценке степени генетической изоляции медведей из разных участков карско-баренцевоморского региона с использованием данных анализа микросателлитных локусов и фрагмента D-петли митохондриальной ДНК.
МАТЕРИАЛЫ И МЕТОДЫ
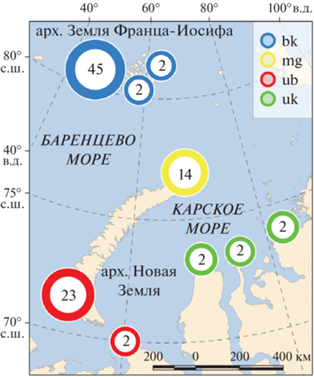
На исследованной территории (архипелаги Земля Франца-Иосифа и Новая Земля, побережье Карского моря от о-ва Вайгач до п-ова Таймыр) были собраны образцы от отловленных и идентифицированных белых медведей (рис. 1). Все манипуляции с животными проводились после химической иммобилизации. Инъекция смеси вводилась с помощью дистанционного пневматического инъектора DanInject JM-25. Забор крови проводился однократно из бедренной вены с помощью одноразового шприца либо вакуумной системы. Во время проведения процедур у всех животных контролировался пульс, оксигенация и температура тела. Далее животному вводили препарат для вывода из наркоза. Данная методика одобрена комиссией по этике ИПЭЭ РАН, протокол № 37 от 25 мая 2020 г.
Рис. 1.
Места сбора проб для генетического анализа: bk – север Баренцева и Карского морей, uk – юг Карского моря, ub – юг Баренцева моря, mg – мыс Желания. Цифры в кружках – объем выборки.
Были проанализированы генетическая структура и генетическое разнообразие группировок животных на разных участках карско-баренцевоморского региона, а также генетическое разнообразие самок и самцов в разных группировках для выявления возможных различий в активности расселения особей за пределы обитания локальной группировки.
Проведен молекулярно-генетический анализ 93 образцов белого медведя, собранных на разных участках карско-баренцевоморского региона. Получены данные по микросателлитным локусам для 89 животных, по митохондриальной ДНК для 93 животных. Информация об использованных в анализе пробах представлена в (табл. 1).
Таблица 1.
Информация об использованных в анализе пробах
| Номер | Пол | Предполагаемая группировка | мтДНК | № NCBI | Микросателлиты |
|---|---|---|---|---|---|
| um003 | m | bk | + | OP923918 | + |
| um5 | m | bk | + | OP923919 | + |
| um6 | f | bk | + | OP923920 | + |
| um7 | f | bk | + | OP923921 | + |
| um8 | m | bk | + | OP923922 | + |
| um9 | f | bk | + | OP923923 | + |
| um10 | f | bk | + | OP923924 | + |
| um12 | m | bk | + | OP923925 | + |
| zfi4 | f | bk | + | OP923926 | + |
| um001 | m | bk | + | OP923933 | + |
| um002 | m | bk | + | OP923934 | + |
| zfi10 | f | bk | + | OP923940 | + |
| zfi11 | f | bk | + | OP923941 | + |
| zfi12 | m | bk | + | OP923942 | + |
| zfi24 | f | bk | + | OP923943 | + |
| zfi25 | m | bk | + | OP923944 | + |
| zfi26 | m | bk | + | OP923945 | + |
| zfi27 | f | bk | + | OP923946 | + |
| zfi28 | m | bk | + | OP923947 | + |
| zfi29 | m | bk | + | OP923948 | + |
| zfi7 | f | bk | + | OP923949 | + |
| zfi8 | m | bk | + | OP923950 | + |
| zfi9 | f | bk | + | OP923951 | + |
| zfi15 | f | bk | + | OP923952 | + |
| zfi16 | f | bk | + | OP923953 | + |
| zfi17 | m | bk | + | OP923954 | + |
| zfi18 | f | bk | + | OP923955 | + |
| zfi19 | m | bk | + | OP923956 | + |
| zfi20 | f | bk | + | OP923957 | + |
| zfi21 | f | bk | + | OP923958 | + |
| zfi22 | f | bk | + | OP923959 | + |
| zfi30 | m | bk | + | OP923984 | + |
| zfi31 | m | bk | + | OP923985 | + |
| zfi32 | m | bk | + | OP923986 | + |
| zfi33 | m | bk | + | OP923987 | + |
| zfi34 | f | bk | + | OP923988 | + |
| zfi35 | f | bk | + | OP923989 | + |
| zfi36 | f | bk | + | OP923990 | + |
| zfi37 | m | bk | + | OP923991 | + |
| zfi38 | m | bk | + | OP923992 | + |
| zfi39 | m | bk | + | OP923998 | + |
| zfi40 | f | bk | + | OP923999 | + |
| zfi41 | f | bk | + | OP924000 | + |
| zfi42 | m | bk | + | OP924001 | + |
| zfi43 | m | bk | + | OP924002 | + |
| um68 | f | bk | + | OP924003 | + |
| zfi3 | f | bk | + | OP923968 | + |
| zfi5 | m | bk | + | OP923992 | + |
| zfi6 | m | bk | + | OP923926 | + |
| nz5 | m | mg | + | OP923964 | + |
| nz6 | m | mg | + | OP923965 | + |
| nz7 | f | mg | + | OP923966 | + |
| nz8 | f | mg | + | OP923967 | + |
| um43 | f | mg | + | OP923977 | + |
| um44 | m | mg | + | OP923978 | + |
| um46 | – | mg | + | OP923979 | – |
| um47 | m | mg | + | OP923982 | – |
| um53 | – | mg | + | OP923983 | – |
| nz9 | m | mg | + | OP923993 | + |
| nz10 | f | mg | + | OP923994 | + |
| nz11 | m | mg | + | OP923995 | + |
| nz12 | m | mg | + | OP923996 | + |
| nz13 | f | mg | + | OP923997 | + |
| bg5 | f | ub | + | OP923927 | + |
| bg2 | f | ub | + | OP923928 | + |
| bg3 | m | ub | + | OP923929 | + |
| bg1 | m | ub | + | OP923930 | + |
| um34 | f | ub | + | OP923935 | + |
| um36 | f | ub | + | OP923936 | + |
| um38 | f | ub | + | OP923937 | + |
| bg4 | f | ub | + | OP923960 | + |
| bg6 | f | ub | + | OP923961 | + |
| bg7 | f | ub | + | OP923962 | + |
| bg8 | m | ub | + | OP923963 | + |
| um55 | m | ub | + | OP923970 | + |
| um58 | m | ub | + | OP923971 | + |
| um59 | f | ub | + | OP923972 | + |
| bg9 | f | ub | + | OP923973 | + |
| bg10 | f | ub | + | OP923974 | + |
| bg11 | m | ub | + | OP923975 | + |
| um56 | f | ub | + | OP923980 | + |
| um57 | m | ub | + | OP923981 | + |
| um39 | m | ub | + | OP923931 | + |
| um40 | f | ub | + | OP923926 | + |
| b_1 | m | ub | + | OP923939 | + |
| b_2 | f | ub | + | OP923926 | + |
| t3 | m | uk | + | OP923931 | + |
| t4 | f | uk | + | OP923932 | + |
| ya3 | f | uk | + | OP923938 | + |
| ya4 | f | uk | + | OP923939 | + |
| ya5 | m | uk | + | OP923968 | + |
| ya6 | f | uk | + | OP923969 | + |
| um30 | m | ub | + | OP923926 | – |
Примечание для табл. 1–4 и 6–8. bk – север Баренцева и Карского морей, mg – мыс Желания, ub – юг Баренцева моря, uk – юг Карского моря.
Образцы крови замораживали в пробирках и транспортировали в замороженном виде или наносили на специальные FTA карты или бинт и высушивали. Остальные образцы консервировали в поваренной соли или 96%-ном этаноле.
ДНК выделяли при помощи набора для выделения DNeasy Blood and Tissue Kit (Qiagen, Германия). Полимеразную цепную реакцию (ПЦР) фрагмента контрольного региона мтДНК проводили с использованием набора Master Mix 5x (Диалат, Россия) и праймеров Bed1 5'-AGCAACAGCTCCACTACCAG-3' и Bed3 5'-CGATTTAGTGGCGTTTATGTAC-3' в следующем режиме: 94°С 3 мин (1 цикл – предварительная денатурация), 94°С 1 мин, 60°С 30 с, 72°С 2 мин (40 циклов), 72°С 5 мин [14]. Очистку продуктов ПЦР проводили методом ферментативной очистки с помощью набора Exo/SAP Go (Grisp, Португалия). Нуклеотидные последовательности определяли с помощью автоматических генетических анализаторов ABI PRISM 3130 и 3500 (Applied Biosystems, США) и применения набора BigDye Terminator v 3.1 (Applied Biosystems). Секвенирование проводили с праймерами, использованными для амплификации. Выравнивание и редактирование последовательностей проводили вручную с помощью программы BioEdit 7.05 [15]. Построение сетей гаплотипов проводилось с помощью программы Network 10.2 [16]. В качестве аутосомных молекулярно-генетических маркеров использовали специально разработанные для медведей 17 микросателлитных локусов из коммерческого набора: COrDIS Bear, включающего 17 пар аутосомных и одну пару половых праймеров (Гордиз, Россия). ПЦР проводили с помощью амплификатора Bio-Rad T 100 (Bio-Rad, США) в объеме 25 мкл, при условиях, рекомендуемых производителем набора. Длины микросателлитных фрагментов определяли на автоматическом генетическом анализаторе Нанофор (Синтол, Россия) и ABI 3500 (Applied Biosystems) с добавлением стандарта длины SD 550 (Гордиз) и программы GeneMapper v 4.1 (Applied Biosystems). Молекулярную изменчивость для микросателлитных локусов AMOVA, соответствие распределения генотипов микросателлитных локусов равновесию Харди–Вайнберга, парные генетические дистанции (Rst) и значения достоверности, ожидаемую гетерозиготность (He) и наблюдаемую гетерозиготность (Ho), гаплотипическое разнообразие (H), нуклеотидную изменчивость (π), парные генетические дистанции (θst) и среднее число аллелей на локус (Na) вычисляли в программе Arlequin 3.5.1.2 [17] и программе Ms tool [18]. Для оценки степени генетической дифференциации между популяциями (θst) использовали молекулярный дисперсионный анализ (AMOVA) с эволюционной моделью Тамура–Неи (TrN + I + G) и значением γ равным 0.8. Эта модель была выявлена как наиболее подходящая для наших данных по информационному критерию Акаике после анализа в программе Modeltest 3.7 [19]. Для тестирования генетической обособленности животных использовали также кластерный анализ Байеса, реализованный в программе Structure 2.3.4 [20], и модель Admicture c коррелированными частотами аллелей для K равного от одного до семи с пятью повторами. Применяли 500 000 повторов и период разогрева 50 000. Для выбора оптимального количества кластеров K, суммирования полученных кластеров и построения графического отображения использовали программу Clumpak [21]. Вероятность наличия нуль-аллелей оценивали в программе Cervus 3.0.3 [22].
РЕЗУЛЬТАТЫ
Анализ митохондриальной ДНК
ДНК успешно выделена из 93 образцов. Для всех образцов получены нуклеотидные последовательности фрагмента контрольного региона мтДНК. Длина выравнивания составила 610 пн. Всего обнаружено 15 вариабельных сайтов и описан 21 гаплотип. Анализировались четыре предполагаемые группы: север Баренцева и Карского морей (bk), юг Карского моря (uk), юг Баренцева моря (ub), мыс Желания (mg) (табл. 1).
Данные выборки анализировали также отдельно для взрослых животных: самцов и самок. Результаты анализа значения генетической дифференциации θst представлены в табл. 2–4.
Таблица 2.
Значения генетической дифференциации θst (под диагональю) и значения достоверности (над диагональю) по данным анализа фрагмента D-петли мтДНК длиной 610 пн для взрослых животных
| Группировки | bk | ub | uk | mg |
|---|---|---|---|---|
| bk | 0 | 0.432 | 0.180 | |
| ub | 0.297* | 0.027 | 0 | |
| uk | 0 | 0.199* | 0.811 | |
| mg | 0.0326 | 0.286* | 0 |
Примечание для табл. 2–4 и 6–8. * Достоверные значения θst (p < 0.05) помечены звездочкой.
Таблица 3.
Значения генетической дифференциации θst (под диагональю) и значения достоверности (над диагональю) по данным анализа фрагмента D-петли мтДНК длиной 610 пн для самок
| Группировки | bk | ub | uk | mg |
|---|---|---|---|---|
| bk | 0 | 0.450 | 0.396 | |
| ub | 0.301* | 0.126 | 0.009 | |
| uk | 0 | 0.101 | 0.676 | |
| mg | 0 | 0.316* | 0 |
Таблица 4.
Значения генетической дифференциации θst (под диагональю) и значения достоверности (над диагональю) по данным анализа фрагмента D-петли мтДНК длиной 610 пн для самцов
| Группировки | bk | ub | uk | mg |
|---|---|---|---|---|
| bk | 0 | 0.901 | 0.477 | |
| ub | 0.245* | 0.216 | 0.018 | |
| uk | 0 | 0.267 | 0.892 | |
| mg | 0 | 0.256* | 0 |
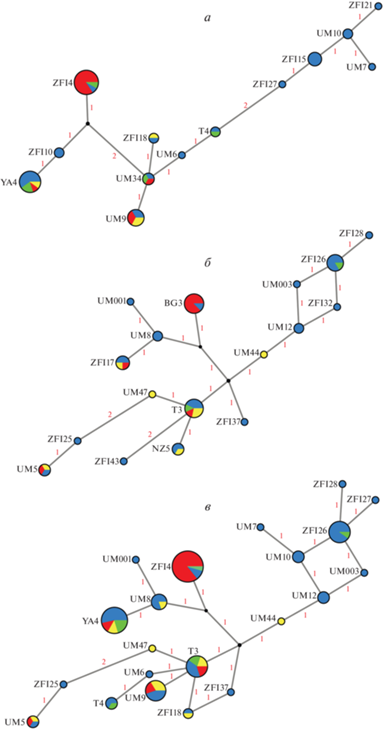
Из-за слабых генетических различий мы объединили выборки bk, uk и mg в одну. При анализе двух группировок достоверное значение θst составило 0.270. При анализе только самцов θst = 0.343, только самок θst = 0.251. Нуклеотидное разнообразие для исследованных выборок составило π (ub) = 0.0031 ± 0.0020, π (bk + uk + mg) = 0.0047 ± ± 0.0028. Гаплотипическое разнообразие H (ub) = = 0.480 ± 0.117, H (bk + uk + mg) = 0.916 ± 0.016. По величине θst группировка юга Баренцева моря (ub) значительно отличается от всех остальных. При этом вклад самцов в эту генетическую изоляцию выше чем самок. Это свидетельствует о том, что взрослые самцы не покидают территории отлова или проводят значительную часть времени на этой территории. Также возможным объяснением может быть эффект основателя и родство животных из этой группировки между собой, так как подавляющее число животных из этой выборки относится к одному гаплотипу. Изменчивость группы и сходство по выбранному митохондриальному маркеру можно оценить по медианной сети гаплотипов (рис. 2) и распределению одинаковых гаплотипов по исследованной территории (рис. 3).
Рис. 2.
Медианная сеть гаплотипов исследованных образцов белого медведя от самок (a), самцов (б) и смешанной выборки (в), построенная при помощи программы Network 10.2 на основании выравнивания нуклеотидных последовательностей контрольного региона мтДНК длиной 610 пн. Длина ветвей пропорциональна количеству мутаций, обозначенных красными цифрами у ветвей. Размер окружностей соответствует числу образцов определенного гаплотипа. Цветом обозначены гаплотипы из различных группировок. Цвета выборок соответствуют рис. 1.
Микросателлитный анализ
Для проведения микросателлитного анализа использована выборка из 89 образцов взрослых белых медведей. По всем 17 локусам для выборки описано 178 аллелей (в среднем 10.47 аллелей на локус). Частоты аллелей исследованных локусов приведены в табл. 5.
Таблица 5.
Частоты аллелей использованных микросателлитных локусов в общей выборке
| Локус | Аллель | Частота | Локус | Аллель | Частота | Локус | Аллель | Частота | Локус | Аллель | Частота | Локус | Аллель | Частота |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7162_072 | 209 | 0.023 | 7204_064 | 265 | 0.011 | 7097_019 | 301 | 0.039 | 7056_179 | 227 | 0.011 | 7031_181 | 339 | 0.035 |
| 213 | 0.148 | 269 | 0.062 | 305 | 0.118 | 231 | 0.017 | 343 | 0.070 | |||||
| 217 | 0.131 | 273 | 0.247 | 309 | 0.270 | 235 | 0.039 | 347 | 0.145 | |||||
| 221 | 0.097 | 277 | 0.230 | 313 | 0.292 | 239 | 0.084 | 351 | 0.180 | |||||
| 225 | 0.244 | 281 | 0.225 | 317 | 0.197 | 243 | 0.225 | 355 | 0.256 | |||||
| 229 | 0.102 | 285 | 0.129 | 321 | 0.051 | 247 | 0.124 | 359 | 0.145 | |||||
| 233 | 0.131 | 289 | 0.045 | 325 | 0.017 | 251 | 0.225 | 363 | 0.105 | |||||
| 237 | 0.102 | 293 | 0.039 | 329 | 0.011 | 255 | 0.140 | 367 | 0.029 | |||||
| 241 | 0.023 | 297 | 0.011 | 337 | 0.006 | 259 | 0.079 | 371 | 0.012 | |||||
| 7027_138 | 251 | 0.017 | 7206_095 | 236 | 0.006 | 7128_106 | 147 | 0.102 | 263 | 0.051 | 375 | 0.017 | ||
| 255 | 0.045 | 240 | 0.006 | 149 | 0.011 | 271 | 0.006 | 379 | 0.006 | |||||
| 259 | 0.079 | 244 | 0.011 | 151 | 0.142 | 7128_139 | 166 | 0.011 | 7200_159 | 183 | 0.039 | |||
| 263 | 0.393 | 248 | 0.067 | 153 | 0.006 | 170 | 0.011 | 187 | 0.022 | |||||
| 267 | 0.146 | 252 | 0.135 | 155 | 0.170 | 174 | 0.219 | 191 | 0.051 | |||||
| 271 | 0.197 | 256 | 0.096 | 159 | 0.244 | 178 | 0.258 | 195 | 0.264 | |||||
| 275 | 0.101 | 260 | 0.163 | 163 | 0.216 | 182 | 0.264 | 199 | 0.348 | |||||
| 279 | 0.017 | 264 | 0.213 | 167 | 0.063 | 186 | 0.163 | 203 | 0.124 | |||||
| 283 | 0.006 | 268 | 0.140 | 171 | 0.028 | 190 | 0.056 | 207 | 0.067 | |||||
| 7126_411 | 285 | 0.035 | 272 | 0.056 | 175 | 0.017 | 194 | 0.011 | 211 | 0.051 | ||||
| 289 | 0.052 | 276 | 0.067 | 7060_084 | 209 | 0.017 | 198 | 0.006 | 215 | 0.022 | ||||
| 293 | 0.140 | 280 | 0.011 | 213 | 0.051 | 7045_229 | 235 | 0.017 | 219 | 0.011 | ||||
| 297 | 0.093 | 284 | 0.022 | 217 | 0.129 | 239 | 0.023 | 7148_319 | 294 | 0.017 | ||||
| 301 | 0.209 | 288 | 0.006 | 221 | 0.197 | 243 | 0.131 | 298 | 0.006 | |||||
| 305 | 0.174 | 7079_531 | 229 | 0.039 | 225 | 0.118 | 247 | 0.256 | 302 | 0.012 | ||||
| 309 | 0.151 | 233 | 0.073 | 229 | 0.163 | 251 | 0.170 | 306 | 0.116 | |||||
| 313 | 0.047 | 237 | 0.146 | 233 | 0.101 | 255 | 0.142 | 310 | 0.099 | |||||
| 317 | 0.058 | 241 | 0.230 | 237 | 0.073 | 259 | 0.085 | 314 | 0.151 | |||||
| 321 | 0.017 | 245 | 0.247 | 241 | 0.039 | 263 | 0.119 | 318 | 0.203 | |||||
| 325 | 0.023 | 249 | 0.174 | 245 | 0.039 | 267 | 0.057 | 322 | 0.174 | |||||
| 7032_021 | 298 | 0.051 | 253 | 0.056 | 249 | 0.051 | 7211_071 | 202 | 0.006 | 326 | 0.070 | |||
| 302 | 0.040 | 257 | 0.011 | 255 | 0.006 | 206 | 0.017 | 330 | 0.081 | |||||
| 306 | 0.136 | 261 | 0.017 | 257 | 0.011 | 210 | 0.185 | 334 | 0.041 | |||||
| 310 | 0.205 | 341 | 0.006 | 261 | 0.006 | 214 | 0.124 | 338 | 0.017 | |||||
| 314 | 0.227 | 218 | 0.298 | 342 | 0.006 | |||||||||
| 318 | 0.136 | 222 | 0.202 | 350 | 0.006 | |||||||||
| 322 | 0.051 | 226 | 0.112 | |||||||||||
| 326 | 0.063 | 230 | 0.056 | |||||||||||
| 330 | 0.045 | |||||||||||||
| 334 | 0.040 | |||||||||||||
| 338 | 0.006 |
Значения генетической дифференциации Rst по данным анализа микросателлитных локусов для взрослых животных (n = 89) представлены в табл. 6–8. Слабую, но достоверную дифференциацию показывают группировки юг Баренцева моря (ub) и север Баренцева и Карского морей (bk). Для выборки самцов и смешанной группы эти различия достоверны, в отличие от выборок самок, что свидетельствует о большем вкладе в изоляцию распределения самцов по исследованной территории.
Таблица 6.
Значения генетической дифференциации Rst (под диагональю) и значения достоверности (над диагональю) по данным анализа 17 микросателлитных локусов для взрослых животных
| Группировки | mg | uk | ub | bk |
|---|---|---|---|---|
| mg | 0.270 | 0.099 | 0.351 | |
| uk | 0.005 | 0.252 | 0.108 | |
| ub | 0.019 | 0.014 | 0 | |
| bk | 0.003 | 0.025 | 0.021* |
Таблица 7.
Значения генетической дифференциации Rst (под диагональю) и значения достоверности (над диагональю) по данным анализа 17 микросателлитных локусов для самок
| Группировки | mg | uk | ub | bk |
|---|---|---|---|---|
| mg | 0.432 | 0.504 | 0.919 | |
| uk | 0 | 0.090 | 0.171 | |
| ub | 0 | 0.039 | 0.081 | |
| bk | 0 | 0.028 | 0.015 |
Таблица 8.
Значения генетической дифференциации Rst (под диагональю) и значения достоверности (над диагональю) по данным анализа 17 микросателлитных локусов для самцов
| Группировки | mg | uk | ub | bk |
|---|---|---|---|---|
| mg | 0.198 | 0.387 | 0.171 | |
| uk | 0.020 | 0.586 | 0.594 | |
| ub | 0.003 | 0 | 0.036 | |
| bk | 0.017 | 0 | 0.026* |
При различных вариантах объединения исследуемых выборок достоверный результат получился только при объединении выборок bk, uk и mg в одну и сравнении ее с выборкой ub. Обе полученные выборки по всем локусам находились в равновесном состоянии генотипов согласно закону Харди–Вайнберга. Небольшое превышение вероятности наличия нуль-аллелей F > 0.05 наблюдается для локуса 7126_411 в выборке bk + uk + mg и для локуса 7045_229 в выборке ub. При анализе двух таких группировок достоверное значение Rst составило 0.018 для смешанной выборки, Rst = 0.022 для самцов и Rst = 0.018 для самок.
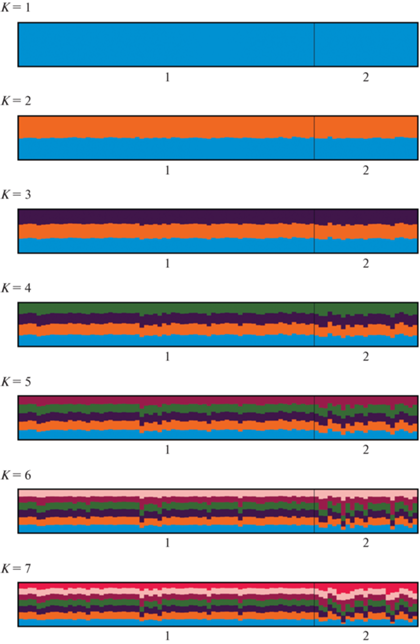
Обработка микросателлитных данных по взрослым животным с помощью кластерного анализа Байеса в программе Structure 2.3.4 с учетом распределения особей по выборкам для наиболее вероятных значений числа генетических кластеров K = 2 не выявила различий в популяционной структуре. Однако минимальному значению log Pr(X|K) соответствует K = 1, что свидетельствует об отсутствии выраженной генетической структуры популяции (рис. 4). Средние значения наблюдаемой и ожидаемой гетерозиготности по всем локусам для выборки ub составляют: Ho = 0.810 ± 0.072, He = 0.823 ± 0.050, для объединенной выборки bk + uk + mg: Ho = 0.834 ± 0.049, He = 0.834 ± 0.035.
Рис. 4.
Генетическая структура популяции белого медведя, исходя из анализа микросателитных данных в программе Structure 2.3.4, где K равно от 1 до 7 с пятью повторами. Для построения использованы 500 000 повторов и период разогрева 50 000.
Мы провели сравнение наших результатов с опубликованными ранее данными по баренцевоморской популяции [11, 23], популяциям моря Бофорта Аляски [24] и Гудзонова залива Канады [25]. Несмотря на то что в цитируемых исследованиях использовались, разные микросателлитные локусы и частично разные фрагменты митохондриальной ДНК, сравнение средних значений ожидаемой (He) и наблюдаемой (Ho) гетерозиготности по всем локусам и гаплотипического разнообразия (Н) дало сравнимые значения. Исключение составляет значительно меньшее значение H для выборки юга Баренцева моря (ub), вероятно из-за родства животных в выборке (табл. 9).
Таблица 9.
Значения индексов генетического разнообразия по литературным и собственным данным. Нуклеотидное разнообразие (π), гаплотипическое разнообразие (H), наблюдаемая гетерозиготность (Ho), ожидаемая гетерозиготность (He), среднее число аллелей на локус Na
| Выборка | H | π | Ho | He | Na |
|---|---|---|---|---|---|
| Популяция юга Баренцева моря (ub) | 0.480 ± 0.117 | 0.0031 ± 0.0020 | 0.810 ± 0.072 | 0.823 ± 0.050 | 7.82 ± 1.70 |
| bk + uk + mg | 0.916 ± 0.016 | 0.0047 ± 0.0028 | 0.834 ± 0.049 | 0.834 ± 0.035 | 10.23 ± 1.92 |
| Популяция севера Баренцева и Карского морей [11, 23] | 0.902 ± 0.014 | 0.0032 ± 0.0018 | 0.61 ± 0.24 | 0.62 ± 0.24 | 8.04 ± 3.07 |
| Популяция моря Бофорта Аляски [24] | – | – | 0.708 | 0.706 | 7.9 |
| Популяция Гудзонова залива Канады [25] | – | – | 0.665 | 0.691 | 7.65 ± 2.17 |
ОБСУЖДЕНИЕ
Анализ генетических данных с использованием микросателлитов и контрольного региона митохондриальной ДНК для количественной оценки генетической дифференциации позволяет выделить в исследованном нами регионе только две популяции (табл. 9). Данные группировки различаются по распределению митохондриального маркера (θst = 0.270) и очень слабо дифференцируются по ядерным локусам (Rst = 0.018), что не позволяет отличать с помощью байесовского кластерного анализа животных из этих группировок. Возможное объяснение генетической дифференциации белых медведей по митохондриальной ДНК на юге архипелага Новая Земля – это влияние антропогенных факторов. При отсутствии возможности охоты на тюленей медведи имеют легкий доступ к большому количеству пищевых отходов человека на свалке ТБО. Результаты двухлетнего мониторинга и мечения белых медведей на архипелаге Новая Земля в районе Белушьей Губы и анализ литературных источников также позволяют предположить, что здесь обитает и размножается отдельная группировка медведей. Изучение перемещений меченых самок и визуальные наблюдения показали, что медведи ежегодно скапливаются в районе свалки ТБО вблизи Белушьей Губы в конце осени в количестве десятков особей и уходят на лед, как только он образуется. Помеченные медведицы зимой и весной не покидали ледовых местообитаний в морских акваториях о-ва Южный и о-ва Вайгач, а после таяния льда много времени проводили в глубине о‑ва Южный. В конце осени они вновь перемещались в район Белушьей Губы [26].
Различия мтДНК, вероятно, отражают как генетический дрейф, так и историческую динамику колонизации территории южной части Баренцева моря. В отличие от этого дифференциация, получаемая с помощью микросателлитов, находится только в масштабе сотен лет и отражает отсутствие современных препятствий для потока генов. Похожие результаты были получены для животных из залива Бутия и пролива Мак-Клинток Канадского Арктического архипелага [27]. Возможно, слабая генетическая дифференциация будет усиливаться при использовании других генетических маркеров – однонуклеотидных замен (SNP) с более низкой изменчивостью и более чувствительных к эффекту основателя. Подтверждение этому можно увидеть в исследовании обширной выборки из Чукотского и Баренцева морей, южной части моря Бофорта и южной части Гудзонова залива. Эти данные указывают на наличие генетической структуризации среди нескольких существующих популяций, расположенных с востока на запад в Полярном бассейне. Fst находился в диапазоне от 0.004 до 0.105 (P < 0.005) среди всех пар популяций, кроме популяций из южной части моря Бофорта и Чукотского моря (Fst = 0.004, P = 0.2). При этом выборки из Баренцева моря (Шпицберген) и южной части Гудзонова залива значительно отличаются от выборок из Аляски [28]. Более сильное воздействие на генетическую изоляцию определяют рельеф и распределение пищевых ресурсов белого медведя. Например, в работе о дифференциации группировки Гудзонова залива с использованием 26 микросателлитных локусов и генетических профилей от 377 белых медведей были выявлены три кластера и значительная их дифференциация по индексу Fst, что позволяет предположить дифференциацию белых медведей, взятых с островов в заливе Джеймс [24]. Эти данные были дополнены и уточнены исследованиями с помощью анализа 2603 однонуклеотидных полиморфизмов. Структура популяций ожидаемо отличалась от микросателлитного исследования. Был выделен дополнительный генетический кластер внутри группировки – бассейн Фокс (FB) [29]. Дальнейшие работы по этому региону уже на 13 488 однонуклеотидных заменах показали соответствие с более ранними работами [30]. Исследование исторических выборок с 1995 по 2016 гг. на микросателлитных данных на архипелаге Шпицберген в Баренцевом море показывает усиление генетической дифференциации на 200% и потерю генетического разнообразия на 3–10% из-за инбридинга отдельных группировок, что, вероятно, связано с изменением ледяного покрова и фрагментацией мест обитания [31]. В нашем исследовании мы также видим большее значение коэффициента инбридинга (Fis = 0.015) для группировки юга Баренцева моря по сравнению с группировкой севера Баренцева и Карского морей (Fis = 0), что также может быть в какой-то степени обусловлено изменением климата и деятельностью человека.
Публикация подготовлена при выполнении работ по анализу и обобщению результатов исследований белого медведя в российской Арктике, проведенных в 2010–2021 гг. Постоянно действующей экспедицией РАН по изучению животных Красной книги Российской Федерации и других особо важных животных фауны России, включенной в состав Института проблем экологии и эволюции им. А.Н. Северцова РАН, в рамках гранта Русского географического общества “Изучение редких видов животных (амурский тигр, дальневосточный леопард, ирбис (снежный барс), белуха, белый медведь)” и в рамках работ по изучению и мониторингу белого медведя и моржа как индикаторов устойчивого состояния морских арктических экосистем по заказу ПАО “НК “Роснефть” (сбор и анализ данных 2020–2021 гг.), а также при поддержке проекта Международного экологического фонда “Чистые моря” в рамках проекта “Хозяин Арктики-2021”.
Авторы благодарят за разработку и возможность использования набора для идентификации медведей российскую компанию “Гордиз”.
Работа выполнена в ЦКП “Инструментальные методы в экологии” ИПЭЭ РАН.
Все применимые международные, национальные и/или институциональные принципы ухода и использования животных были соблюдены.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Liu S., Lorenzen E.D., Fumagalli M. et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears // Cell. 2014. V. 157. P. 785–794. https://doi.org/10.1016/j.cell.2014.03.054
Hassanin A. The role of Pleistocene glaciations in shaping the evolution of polar and brown bears. Evidence from a critical review of mitochondrial and nuclear genome analyses // C. R. Biol. 2015. V. 338. P. 494–501.https://doi.org/10.1016/j.crvi.2015.04.008
Taylor M.K., Akeeagok S., Andriashek D. et al. Delineating Canadian and Greenland polar bear (Ursus maritimus) populations by cluster analysis of movements // Can. J. Zool. 2001. V. 79. P. 690–709. https://doi.org/10.1139/z01-028
Paetkau D., Amstrup S.C., Born E.W. et al. Genetic structure of the world’s polar bear populations // Mol. Ecol. 1999. V. 8. P. 1571–1584. https://doi.org/10.1046/j.1365-294x.1999.00733.x
Malenfant R.M., Coltman D.W., Davis C.S. Design of a 9K Illumina BeadChip for polar bears (Ursus maritimus) from RAD and transcriptome sequencing // Mol. Ecol. Resour. 2015. V. 15(3). P. 587–600. https://doi.org/10.1111/1755-0998.12327
Cronin M.A., Amstrup S.C., Scribner K.T. Microsatellite DNA and mitochondrial DNA variation in polar bears in the Beaufort and Chukchi seas, Alaska. // Can. J. Zool. 2006. № 84. P. 655–660. https://doi.org/10.1139/Z06-039
Peacock E., Sonsthagen S.A., Obbard M.E. et al. Implications of the circumpolar genetic structure of polar bears for their conservation in a rapidly warming arctic // PLoS One. 2015 V. 10(1). https://doi.org/10.1371/journal.pone.0112021
Malenfant R.M., Davis C.S., Cullingham C.I., Coltman D.W. Circumpolar genetic structure and recent gene flow of polar bears: a reanalysis // PLoS One. 2016. V. 11(3). https://doi.org/10.1371/journal.pone.0148967
Mauritzen M., Derocher A.E., Wiig Ø. et al. Using satellite telemetry to define spatial population structure in polar bears in the Norwegian and western Russian Arctic // J. Appl. Ecol. 2002. V. 39. P. 79–90. https://doi.org/10.1046/j.1365-2664.2002.00690.x
Amstrup S.C., Durner G.M., McDonald T.L. et al. Comparing movement patterns of satellite-tagged male and female polar bears // Can. J. Zool. 2001. V. 79. P. 2147–2158. https://doi.org/10.1139/z01-174
Zeyl E., Ehrich D., Aars J. et al. Denning-area fidelity and mitochondrial DNA diversity of female polar bears (Ursus maritimus) in the Barents Sea // Can. J. Zool. 2010. V. 88. P. 1139–1148. https://doi.org/10.1139/Z10-078
Laidre K., Born E., Gurarie E. et al. Females roam while males patrol: Divergence in breeding season movements of pack-ice polar bears (Ursus maritimus) // Proc. Biol. Sciences. The Royal Society. 2013. V. 280. https://doi.org/10.1098/rspb.2012.2371
Wiig Ø., Born E., Laidre K. et al. Performance and retention of lightweight satellite radio tags applied to the ears of polar bears (Ursus maritimus) // Animal Biotelemetry. 2017. V. 5. P. 1–11. https://doi.org/10.1186/s40317-017-0124-0
Matsuhashi T., Masuda R., Mano T. et al. Microevolution of the mitochondrial DNA control region in the japanese brown bear (Ursus arctos) population // Mol. Biol. Evol. 1999. V. 16(5). P. 676–684. https://doi.org/10.1093/oxfordjournals.molbev.a026150
Hall T.A. Bioedit: A user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT // Nucl. Acids Symp. Series. 1999. V. 41. P. 95–98.
Bandelt H.J., Forster P., Rohl A. Median-Joining networks for inferring intraspecific phylogenies // Mol. Biol. Evol. 1999. V. 16. № 1. P. 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Excoffier L.G., Lischer H.E. Arlequin suite ver 3.5: А new series of programs to perform population genetics analyses under Linux and Windows // Mol. Ecol. Resour. 2010. V. 10. P. 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Park S.D.E. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Ph.D. thesis Univ. Dublin, 2001. 53 p.
Posada D., Crandall K.A. Modeltest: Testing the model of DNA substitution // Bioinformatics. 1998. V. 14. № 9. P. 817–818. https://doi.org/10.1093/bioinformatics/14.9.817
Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data // Genetics. 2000. V. 155. P. 945–959. https://doi.org/10.1093/genetics/155.2.945
Kopelman N.M., Mayzel J., Jakobsson M. et al. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K // Mol. Ecol. Resour. 2015. V. 15(5). P. 1179–1191. https://doi.org/10.1111/1755-0998.12387
Kalinowski S.T., Taper M.L., Marshall T.C. et al. Revising how the computer program CERVUS accommodates genotyping error increases success inpaternity assignmen // Mol. Ecol. 2007. V. 16. P. 1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Zeyl E., Aars J., Ehrich D., Wiig Ø. Families in space: Relatedness in the Barents sea population of polar bears (Ursus maritimus) // Mol. Ecol. 2009. V. 18(4). P. 735–749. https://doi.org/10.1111/j.1365-294X.2008.04049.x
Cronin M.A., Amstrup S.C., Talbot S.L. et al. Genetic variation, relatedness, and effective population size of polar bears (Ursus maritimus) in the southern Beaufort sea, Alaska // J. Hered. 2009. № 100. P. 681–690. https://doi.org/10.1093/jhered/esp061
Crompton A.E., Obbard M.E., Petersen S.D., Wilson P.J. Population genetic structure in polar bears (Ursus maritimus) from Hudson Bay, Canada: implications of future climate change // Biol. Conserv. 2008. V. 141(10). P. 2528–2539. https://doi.org/10.1016/j.biocon.2008.07.018
Платонов Н.Г., Мизин И.А., Иванов Е.А. и др. Использование белым медведем (Ursus maritimus) местообитаний вдоль береговой линии в течение года по данным спутникового мониторинга // Исследование Земли из космоса. 2019. № 3. С. 80–91. https://doi.org/10.31857/S0205-96142019380-91
Campagna L., Van Coeverden de Groot P.J., Saunders B.L. Extensive sampling of polar bears (Ursus maritimus) in the Northwest Passage (Canadian Arctic Archipelago) reveals population differentiation across multiple spatial and temporal scales // Ecol. Evol. 2013. V. 3(9). P. 3152–3165. https://doi.org/10.1002/ece3.662
Miller W., Schuster S.C., Welch A.J. et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change // Proc. Natl. Acad. Sci. USA. 2012. V. 109(36). P. E2382–E2390. https://doi.org/10.1073/pnas.1210506109
Viengkone M., Derocher A.E., Richardson E.S. et al. Assessing polar bear (Ursus maritimus) population structure in the Hudson Bay region using SNPs // Ecol. Evol. 2016. V. 6. P. 8474–8484. https://doi.org/10.1002/ece3.2563
Jensen E.L., Tschritter C., de Groot P.V.C. et al. Canadian polar bear population structure using genome-wide markers // Ecol. Evol. 2020. P. 1–9. https://doi.org/10.1002/ece3.615
Maduna S., Aars J., Fløystad I. et al. Sea ice reduction drives genetic differentiation among Barents sea polar bears // Proc. R. Soc. B. 2021. https://doi.org/10.1098/rspb.2021.1741
Дополнительные материалы отсутствуют.