

Известия РАН. Серия биологическая, 2019, № 2, стр. 197-203
Сопряженность сократительной активности миокарда и уровня окислительного стресса у крыс при сочетанном развитии постинфарктного кардиосклероза и сахарного диабета
Д. С. Кондратьева *, С. А. Афанасьев, Т. Ю. Реброва, С. В. Попов
Научно-исследовательский институт кардиологии, Томский национальный медицинский центр РАН
634012 Томск, ул. Киевская, 111а, Россия
* E-mail: dina@cardio-tomsk.ru
Поступила в редакцию 21.01.2016
После доработки 28.05.2018
Принята к публикации 27.04.2018
Аннотация
Исследованы особенность функционального состояния миокарда крыс и активность процессов перекисного окисления липидов при сочетанном развитии сердечной недостаточности ишемического генеза и сахарного диабета. Оценены ритмоинотропная реакция миокарда и содержание активных продуктов тиобарбируровой кислоты и диеновых конъюгатов в плазме крови крыс с постинфарктным и диабетическим ремоделированием сердца, а также при их сочетанном развитии. Обнаружено, что в условиях эксперимента при формировании постинфарктного ремоделирования сердца индукция сахарного диабета приводит к сдерживанию роста активности процессов перекисного окисления липидов и сохранению ритмоинотропных реакций миокарда.
Хорошо известно, что метаболические нарушения, развивающиеся при сахарном диабете (СД), усугубляют развитие сердечной недостаточности (СН) ( Elder et al., 2016). В значительной мере это обусловлено изменением энергетического метаболизма. При этом диабетическая кардиомиопатия сопровождается ремоделированием мембран кардиомиоцитов конечными продуктами гликирования и свободнорадикального окисления (Ziegelhöffer-Mihalovicová et al., 2003; Ziegelhöffer et al., 2012). Все это способствует нарушению электрической стабильности мембран и ионного баланса клеток сердца. Ключевая структура, отвечающая за внутриклеточный транспорт Са2+ и за инотропный ответ кардиомиоцитов, – саркоплазматический ретикулум (СР). Установлена взаимосвязь между изменением гомеостаза Са2+ в кардиомиоцитах и прогрессированием СН: нарушение внутриклеточного транспорта Са2+ предшествует депрессии механической работы сердца (Lehnart et al., 2009; Lou et al., 2011).
Развитие и СН, и СД сопровождается значительным повышением активности перекисного окисления липидов (ПОЛ) (Tsutsui et al., 2011; Wu et al., 2013). Продукты ПОЛ увеличивают проницаемость липидной фазы мембран для ионов водорода и кальция. В митохондриях это приводит к разобщению процессов окисления и фосфорилирования, и клетка оказывается в условиях энергетического голодания. При этом в цитоплазму поступает избыточное количество Са2+, которое способствует повреждению клеточных структур.
Если клинические данные однозначно указывают на снижение устойчивости диабетического сердца к действию ишемии, то результаты экспериментальных исследований достаточно противоречивы. Так, in vivo и in vitro отмечена парадоксально высокая ишемическая резистентность миокарда животных с небольшим сроком стрептозотоцининдуцированного диабета (Chen et al., 2006; Ravingerová et al., 2010). В наших предварительных исследованиях также были обнаружены факты сохранения сократительной активности миокарда при сочетанном моделировании СН и СД. Механизм этого феномена остается предметом научного поиска.
Цель работы – исследовать особенность функционального состояния миокарда крыс и активности ПОЛ при сочетанном развитии СН ишемического генеза и СД.
МАТЕРИАЛЫ И МЕТОДЫ
Работа выполнена на половозрелых крысах-самцах линии Wistar массой 200–220 г. Были сформированы четыре группы животных: I – интактные крысы (n = 12), II – крысы с постинфарктным кардиосклерозом (ПИКС) (n = 11), III – крысы с индуцированным СД (n = 8), IV – крысы, которым через 2 нед после коронароокклюзии индуцировали СД (n = 8). Животных подбирали таким образом, чтобы на момент эксперимента возраст крыс во всех группах был одинаковый. Инфаркт миокарда моделировали путем окклюзии левой нисходящей коронарной артерии (Кондратьева и др., 2013), после чего животных содержали в стандартных условиях вивария. Диабет моделировали однократным введением внутрибрюшинно стрептозотоцина (Sigma, США) в дозе 60 мг/кг, разведенного ex tempore 0.01 моль/л цитратным буфером (рН 4.5). С крысами IV группы проводили эксперименты через 6 нед после индукции диабета. Концентрацию глюкозы в сыворотке крови определяли с помощью ферментно-колориметрического теста (Biocon Diagnostic, Германия).
Развитие гипертрофии сердца и левого желудочка (ЛЖ) оценивали по соотношению масс сердца и тела животного и отношению масс ЛЖ и сердца (Satoh et al., 2001). Размер постинфарктных рубцов сердца животных оценивали методом планиметрии и рассчитывали в процентах площади общей стенки ЛЖ (Усачева и др., 2007).
В день эксперимента у животных забирали кровь в пробирку с гепарином (10 : 1). Пробы крови центрифугировали при 3000 об./мин в течение 5 мин. Полученную плазму разливали на аликвоты и хранили в жидком азоте до момента исследования. Активность ПОЛ в плазме крови оценивали с использованием ранее описанных (Реброва и др., 2007) биохимических методов определения содержания продуктов, образующихся при свободнорадикальном окислении липидов, в реакции с 2-тиобарбитуровой кислотой (ТБК), измеряя концентрацию ТБК-активных продуктов (ТБК-АП). Так же спектрофотометрически оценивали содержание диеновых конъюгатов (ДК) в гексановых экстрактах.
Сократительную активность изучали на папиллярных мышцах. Для этого животных, находящихся под рауш-наркозом, обездвиживали смещением шейного отдела позвоночника и вскрывали грудную клетку. Выделенное сердце промывали в специализированной проточной камере через аорту раствором Кребса–Хензеляйта следующего состава, мМ: NaCl – 120, KCl – 4.8, CaCl2 – 2, Mg2SO4 – 1.2, KH2PO4 – 1.2, NaHCO3 – 20.0, глюкоза – 10 (Sigma). После этого выделяли папиллярные мышцы и помещали их в термостабилизированную (36°С) проточную камеру. Перфузию мышц осуществляли раствором Кребса–Хензеляйта. Оксигенацию раствора проводили карбогеном (О2 – 95%, СО2 – 5%). Сократительную активность мышц оценивали в изометрическом режиме, используя датчик Force transducer, KG-Series (Scientific Instruments GmbH, Германия). Напряжение, развиваемое мышцей, оценивали в пересчете на площадь поперечного сечения изолированной мышцы (мН/мм2). Стимуляцию мышц проводили электрическими импульсами прямоугольной формы длительностью 5 мс с частотой 0.5 Гц. Перед началом исследования мышцы адаптировали к условиям перфузии и изометрическому режиму в течение 60 мин.
Известно, что функциональное состояние изолированных полосок миокарда можно оценивать, изменяя режим их электрической стимуляции. При экстрасистолических (ЭС) воздействиях регистрируют ЭС-сокращение, которое характеризует возбудимость сарколеммы (Vassallo et al., 1995), и постэкстрасистолическое (ПЭС) сокращение, которое отражает способность СР кардиомиоцитов аккумулировать Са2+, дополнительно поступающие в миоплазму при внеочередном возбуждении и определяющие амплитуду сокращения (Vassallo et al., 1995). В нашей работе ЭС-воздействие оказывали дополнительным электрическим импульсом, наносимым однократно через 0.2, 0.225, 0.25, 0.5, 0.75, 1 и 1.5 с (ЭС-интервал) от начала регулярного цикла. Амплитуду ЭС- и ПЭС-сокращений выражали в процентах амплитуды регулярного (базового) цикла. Анализировали зависимость изменений амплитуды ЭС- и ПЭС-сокращений от длительности ЭС-интервала.
Данные исследований представлены в таблицах в виде Х ± х (Х – среднее значение показателя, х – статистическая ошибка среднего), а также Me (25, 75) (медианы и процентилей). Достоверность полученных данных оценивали с помощью двустороннего t-критерия Стьюдента и непараметрического критерия Манна–Уитни (U). Статистически значимыми считали различия при р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Результаты, полученные в ходе исследования, показали, что у животных к концу формирования постинфарктного кардиосклероза (II группа) масса тела была меньше таковой у интактных животных на 18.8% (табл. 1). Ремоделирование сердца этих животных сопровождалось развитием гипертрофии миокарда. Так, соотношение масс сердца и тела крыс с ПИКС превышало соответствующее соотношение масс сердца и тела интактных животных на 91% (р < 0.05). Индукция диабета (III группа) приводила к снижению массы тела животных на 56% (р < 0.05), но при этом гипертрофия сердца не развивалась. При сочетанном формировании ПИКС с СД (IV группа) масса тела животных снижалась на 26% по сравнению с таковой в I группе. У крыс с сочетанной патологией, аналогичной таковой в группе III, гипертрофия сердца не развивалась. При этом оказалось, что и размеры зоны рубца ЛЖ в группах II и IV достоверно не различались. Концентрация глюкозы в крови животных III и IV групп превышала таковую у интактных крыс в 4.5 и 3 раза соответственно, что свидетельствует о возникновении стойкой хронической гипергликемии у животных с индуцированным СД.
Таблица 1.
Масса тела, отношение массы сердца и тела и содержание глюкозы в крови крыс с постинфарктным кардиосклерозом и сахарным диабетом
| Группа | n | Масса тела, г | Глюкоза, М/л | Масса сердца/масса тела, мг/г | Масса ЛЖ/масса сердца, мг/мг | Зона рубца, % |
|---|---|---|---|---|---|---|
| I | 12 | 298 ± 23.7 | 6 ± 0.37 | 3.29 ± 0.21 | 0.645 ± 0.013 | – |
| II | 11 | 242 ± 11.2* | 7 ± 0.13 | 6.27 ± 0.33* | 0.687 ± 0.02* | 51.3 ± 8.9 |
| III | 8 | 160 ± 14.8*# | 27 ± 2.8*# | 3.77 ± 0.31# | 0.654 ± 0.01 | – |
| IV | 8 | 221 ± 4.5*# | 18 ± 1.8*# | 3.37 ± 0.11# | 0.663 ± 0.02 | 46.1 ± 2.7 |
В нашем исследовании ремоделирование миокарда как после стенозирования коронарной артерии (II группа), так и после развития гипергликемии (III группа) приводило к изменению инотропной реакции папиллярных мышц на ЭС-воздействия по сравнению с контролем (рис. 1а). Так, амплитуда ЭС-сокращений папиллярных мышц крыс с ПИКС (II группа) на коротких ЭС-интервалах была выше таковой у интактных животных на 8% (р < 0.05). После самого длинного ЭС-интервала эта разница достигала 16% (р < 0.05). ЭС-сокращения папиллярных мышц крыс III группы имели свои особенности. Так, самостоятельное ЭС-сокращение появлялось уже при ЭС-интервале 0.225 с, тогда как в остальных группах ЭС-сокращение возникало только при воздействии электрическим импульсом через 0.25 с от начала регулярного цикла. Кроме того, в III группе на коротких ЭС-интервалах амплитуда ЭС-сокращения была на 20% выше, чем в I группе (интактные животные). При воздействии электрическим импульсом после длинных ЭС-интервалов эта разница уменьшалась до 7% (рис. 1а). Исследование инотропного ответа миокарда животных IV группы на ЭС-воздействия показало, что амплитуда ЭС-сокращений достоверно не отличалась от таковой у интактных крыс (I группа).
Рис. 1.
Экстрасистолические (а) и постэкстрасистолические (б) сокращения миокарда крыс с постинфарктным кардиосклерозом (ПИКС) и сахарным диабетом (СД). I – группа интактных крыс, II – группа крыс с ПИКС, III – группа крыс с СД, IV – группа крыс с ПИКС и СД. * – р < 0.05 (статистически значимые различия по сравнению с I группой), # – р < 0.05 (статистически значимые различия по сравнению со II группой), ^ – р < 0.05 (статистически значимые различия по сравнению с III группой).
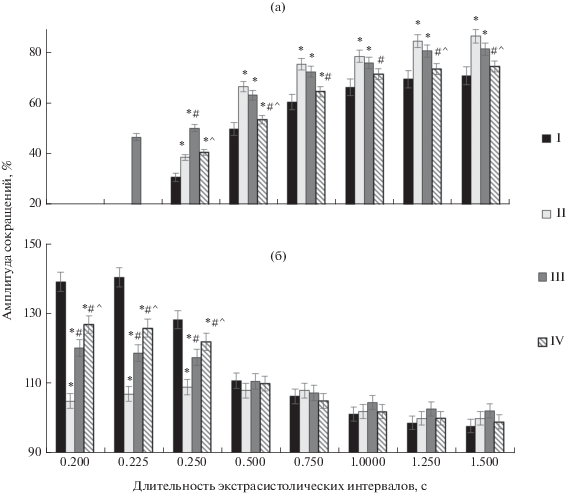
Внеочередной импульс электрической стимуляции при ЭС-интервале 0.2 с не вызывал ЭС-сокращения папиллярных мышц интактных крыс (I группа), но при этом наблюдалось повышение амплитуды ПЭС-сокращения на 39% по сравнению с амплитудой регулярного цикла (рис. 1б). С появлением ЭС-сокращения (при интервале 0.25 с) и повышением его амплитуды наблюдалось снижение амплитуды ПЭС-сокращения. Увеличение длительности ЭС-интервалов приводило к снижению потенциации сокращения после ЭС-воздействия, и в группе интактных животных на самых длинных ЭС-интервалах потенциация ПЭС-сокращения отсутствовала (рис. 1б).
Как видно на рис. 1б, у крыс в группе II потенциация ПЭС папиллярных мышц практически не наблюдалась независимо от длительности ЭС-интервала. Этот факт может свидетельствовать о значительном снижении Са2+-депонирующей функции СР. При исследовании папиллярных мышц крыс III группы потенциация ПЭС-сокращения была значительно меньше таковой у крыс I группы (интактные красы) и составляла 21–16% (рис. 1б). Ремоделирование миокарда при сочетанном развитии постинфарктного и диабетического поражения (IV группа) характеризовалось тем, что повышение ПЭС-сокращения папиллярных мышц составляло 27–19% на коротких ЭС-интервалах (рис. 1б). Сохранение ПЭС-потенциации папиллярных мышц у крыс с сочетанной патологией может свидетельствовать о лучшем сохранении Са2+-депонирующей способности СР, чем у животных с монопатологией.
В табл. 2 представлены данные, полученные при определении концентрации продуктов ПОЛ в плазме крови крыс экспериментальных групп. По окончании формирования ПИКС у крыс II группы было отмечено достоверное увеличение содержания ДК (в 1.9 раза) и ТБК-АП (в 2.6 раза) по отношению к соответствующим показателям в группе интактных крыс. Моделирование СД (III группа) способствовало более выраженному повышению показателей активности протекания свободнорадикальных процессов в плазме крови. В образцах плазмы животных этой группы было отмечено превышение концентрациями ДК в 3.7 раза и ТБК-АП в 2.1 раза аналогичных показателей в группе интактных животных. При этом увеличение наработки ДК было достоверно выше (в 1.9 раза) по сравнению с таковыми у крыс II группы (ПИКС). Значения концентрации ТБК-АП в плазме крови животных II и III групп достоверно не различались.
Таблица 2.
Содержание диеновых конъюгатов (ДК) и тиобарбитуровой кислоты – активных продуктов (ТБК-АП) в плазме крови у животных экспериментальных групп (Me (Q1;Q3))
| Группы | n | ДК, ΔЕ232/ мл | ТБК-АП, ммоль/л |
|---|---|---|---|
| I | 12 | 0.34 (0.16, 0.38) | 10.25 ( 7.59, 14.2 ) |
| II | 11 | 0.66 (0.62, 0.74)* | 27.23 (26.25, 28.65)* |
| III | 8 | 1.26 (0.8, 1.68)*# | 21.22 (13.23, 35.53) |
| IV | 8 | 0.92 (0.6, 1.18)* | 14.72 (13.23, 16.61)# |
Сочетанное развитие ПИКС и СД у крыс IV группы теоретически должно было сопровождаться более выраженной активацией ПОЛ, однако мы получили парадоксальной результат. Так, среднее значение ТБК-АП в этой группе оказалось достоверно ниже (в 1.8 раза), чем во II группе. Достоверных различий при сравнении значений ТБК-АП в группах III и IV мы не обнаружили. Среднее значение ДК в группе с сочетанной патологией было выше такового во II группе, но ниже такового в III группе. Однако эти изменения не были статистически значимыми.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Моделирование СД с помощью стрептозотоцина приводит к стойкой хронической гипергликемии и существенному снижению массы тела животных. Эти показатели подтверждают развитие у крыс СД. Вместе с тем, как оказалось, состояние животных при моделировании сочетанной патологии было значительно лучше состояния животных с монопатологией. Так, снижение массы тела было менее выражено, хотя и статистически значимо ниже по сравнению с массами тела контрольных крыс. При этом содержание глюкозы в крови хотя и оставался высоким, но было на 33% ниже такового у животных с СД. Эти данные свидетельствуют о том, что у животных с сочетанной патологией СД также развивался. Моделирование инфаркта миокарда у крыс перед индукцией СД приводило к формированию соединительно-тканного рубца, размер которого был почти таким же, как и у животных с ПИКС. Эти результаты подтверждают, что у животных с сочетанной патологией наблюдались признаки как постинфарктного кардиосклероза, так и СД.
Исследование сократительной активности миокарда у животных рассматриваемых групп показало, что при ПИКС и СД возбудимость миокарда повышается, о чем свидетельствует повышение амплитуды ЭС-сокращений. Увеличение силы сокращения мышцы возможно в результате увеличения количества Са2+, участвующего в ЭС-сокращении, или/и повышения чувствительности миофиламентов кардиомиоцитов к Са2+. Показано, что ишемическое поражение сердца характеризуется угнетением АТФ-зависимых процессов, в том числе внутриклеточных ион-транспортирующих систем, которое вызывает повышение внутриклеточной концентрации Na+ и Са2+ (Hansen et al., 2007; Hund et al., 2008; Decker, Rudy, 2010) и, соответственно, приводит к увеличению возбудимости мембран кардиомиоцитов. При этом у животных с СД возбудимость повышается в большей степени, поскольку ЭС-сокращение возникает на более раннем ЭС-интервале, чем у животных других групп. Известно, что ЭС-воздействие вызывает инотропный ответ только в том случае, если оно попадает в фазу относительной рефрактерности (Vassallo et al., 1995). Можно отметить, что развитие СД ведет к укорочению фазы абсолютной рефрактерности, а значит вызывает повышение возбудимости кардиомиоцитов. Однако при формировании сочетанной патологии параметры возбудимости и структуры ЭС-сокращений сохранялись на уровне таковых интактных животных.
Известно, что стимул, наносимый во время третьей фазы (абсолютный рефрактерный период) потенциала действия кардиомиоцитов, не способен вызвать сократительный ответ. Однако он инициирует дополнительное поступление внешних ионов кальция в миоплазму. Эти дополнительные Са2+ аккумулируются в СР и участвуют в первом ПЭС-сокращении (Vassallo et al., 1995). В связи с этим амплитуда ПЭС-сокращения превышает амплитуду регулярного цикла интактного миокарда. Однако на фоне формирования постинфарктного кардиосклероза потенциация ПЭС-сокращения миокарда не наблюдалась. Вероятно, в условиях постинфарктного ремоделирования миокарда крыс в кардиомиоцитах нарушается работа Са2+-транспортирующих систем, сопряженных с СР, что приводит к снижению количества ионов кальция во внутриклеточном депо (Lehnart et al., 2009; Lou et al., 2011). На фоне сахарного диабета ПЭС-потенциация сохранялась, хотя значения ее были ниже таковых у контрольных животных. В экспериментальных исследованиях на моделях диабета I типа было обнаружено снижение активности и/или экспрессии SERCA2a уже на ранней стадии развития диабета. Такое изменение функциональной активности этого белка приводит к снижению кальций-аккумулирующей функции СР и, соответственно, к нарушению расслабления кардиомиоцитов (Zhong et al., 2001). На поздней стадии развития СД I типа у крыс значительно снижается число рианодиновых рецепторов, что приводит к серьезным дефектам систолической функции кардиомиоцитов (Netticadan et al., 2001; Zhong et al., 2001). Вместе с тем при формировании сочетанной патологии потенциация ПЭС-сокращения была достоверно выше таковой у животных с СД и ПИКС. Эти результаты показывают, что в случае сочетанной патологии функциональная активность СР сохраняется.
Известно, что повышенная активность ПОЛ – важный компонент повреждения кардиомиоцитов при инфаркте миокарда (Misra et al., 2009) и СД (Wu et al., 2013). Повреждение липидного бислоя мембран радикалами кислорода рассматривается как один из механизмов нарушения внутриклеточного гомеостаза Са2+ и сократительной активности кардиомиоцитов. Ранее мы показали, что повышенная активность ПОЛ сохраняется и при постинфарктном ремоделировании сердца. Более того, при моделировании ПИКС-динамики изменений содержания продуктов ПОЛ (TБК-AП и ДК) в ткани миокарда и плазме крови крыс совпадали (Реброва и др., 2007). В нашем исследовании моделирование протекания ПИКС или СД приводит к повышению активности ПОЛ. При этом при СД в большей степени активируются начальные этапы цепи реакций ПОЛ с образованием ДК, а при ПИКС преобладают заключительные реакции каскада процессов ПОЛ. Активные формы кислорода в патологически высоких концентрациях вступают в реакцию как с липидами, так и с белками клеточных мембран и компонентов плазмы крови. В литературе встречаются данные о снижении активности белков-ферментов, в том числе и Са2+-АТФазы кардиомиоцитов (Köhler et al., 2014) при патологиях, сопровождающихся активацией свободнорадикальных процессов. Возможно, на фоне хронической ишемии миокарда и гипергликемии избыточная активация свободнорадикальных процессов вызывает структурные повреждения клеточных органелл и, как следствие, снижение активности Са2+-АТФазы СР (Köhler et al., 2014), а нарушение целостности липидного бислоя мембран может способствовать утечке Са2+ из СР. Сочетанное развитие патологических состояний приводит к уменьшению концентрации ТБК-АП, что свидетельствует о снижении интенсивности протекания заключительных этапов реакций ПОЛ. Факт незначительного снижения ДК свидетельствует о том, что интенсивность начальных этапов ПОЛ остается достаточно высокoй.
Наши данные свидетельствуют, что индукция диабета на фоне формирования постинфарктного ремоделирования парадоксально способствует сохранению функциональной активности Са2+-транспортирующих систем, сопряженных с СР, и снижению активности ПОЛ. Возможно, это связано с тем, что на фоне развивающейся гипергликемии продукты гликозилирования увеличивают ригидность мембран кардиомиоцитов (Waczulikova et al., 2002). Усиление адаптивных реакций при сочетанном развитии постинфарктных и диабетических нарушений миокарда может быть связано с особенностями внутриклеточного энергетического метаболизма при данных патологических состояниях. Так, повышение содержания глюкозы на начальных стадиях развития ПИКС позволяет активизировать процессы гликолиза в кардиомиоцитах. Известно, что положительное влияние глюкозы на работу сердца при экспериментальной ишемии миокарда связано с повышением гликолитической продукции АТФ (Ardehali et al., 2012; Doenst et al., 2013). Сдвиг энергетического метаболизма в сторону гликолитической продукции АТФ сохраняет функциональную активность Са2+-транспортирующей системы СР при сочетанной патологии. Это согласуется с тем, что АТФ, образующаяся в процессе гликолиза – незаменимый источник энергии для Са2+-транспортирующей системы СР (Zima et al., 2006). Полученные нами данные согласуются с результатами других исследований, свидетельствующих об ишемической резистентности миокарда (in vivo и in vitro) животных с небольшим сроком стрептозотоцининдуцированного диабета (Nawata et al., 2002; Chen et al., 2006).
Таким образом, результаты исследований показали, что в условиях эксперимента, индукция СД на стадии формирования постинфарктного ремоделирования повышает адаптивные возможности миокарда. Это проявляется в сдерживании роста активности ПОЛ и сохранении ритмоинотропных реакций миокарда, связанных с работой Са2+-транспортирующих систем СР.
Работа выполнена в рамках темы фундаментальных исследований № АААА-А15-115123110026-3.
Список литературы
Кондратьева Д.С., Афанасьев С.А., Попов С.В. Экспрессия Са2+-АТФазы саркоплазматического ретикулума кардиомиоцитов крыс при экспериментальном постинфарктном кардиосклерозе и сахарном диабете // Бюл. эксперим. биологии и медицины. 2013. Т. 156. № 12. С. 709–712.
Реброва Т.Ю., Кондратьева Д.С., Афанасьев С.А., Барзах Е.И. Активность перекисного окисления липидов и функциональное состояние миокарда при ремоделировании сердца крыс после экспериментального инфаркта // Кардиология. 2007. № 6. С. 41–45.
Усачева М.А., Попкова Е.В., Смирнова Е.А., Салтыкова В.А., Белкина Л.М. Адаптация сердечно-сосудистой системы к постинфарктному кардиосклерозу у крыс с разной врожденной адренореактивностью миокарда // Бюл. эксперим. биологии и медицины. 2007. Т. 144. № 12. С. 624–628.
Ardehali H., Sabbah H.N., Burke M.A., Sarma S., Liu P.P., Cleland J.G., Maggioni A., Fonarow G.C., Abel E.D., Campia U., Gheorghiade M. Targeting myocardial substrate metabolism in heart failure: potential for new therapies // Eur. J. Heart Fail. 2012. V. 14(2). P. 120–129.
Chen H., Shen W.L., Wang X.H., Chen H.Z., Gu J.Z., Fu J., Ni Y.F., Gao P.J., Zhu D.L., Higashino H. Paradoxically enhanced heart tolerance to ischaemia in type 1 diabetes and role of increased osmolarity // Clin. Exp. Pharmacol. Physiol. 2006. V. 33(10). P. 910–916.
Decker K.F., Rudy Y. Ionic mechanisms of electrophysiological heterogeneity and conduction block in the infarct border zone // Am. J. Physiol. Heart Circ. Physiol. 2010. V. 299(5). P. H1588–H1597.
Doenst T., Nguyen T.D., Abel E.D. Cardiac metabolism in heart failure: implications beyond ATP production // Circ. Res. 2013. V. 113(6). P. 709–724.
Elder D.H., Singh J.S., Levin D., Donnelly L.A., Choy A.M., George J., Struthers A.D., Doney A.S., Lang C.C. Mean HbA1c and mortality in diabetic individuals with heart failure: a population cohort study // Eur. J. Heart Fail. 2016. V. 18(1). P. 94–102.
Hansen P.S., Clarke R.J., Buhagiar K.A., Hamilton E., Garcia A., White C., Rasmussen H.H. Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes // Am. J. Physiol. Cell Physiol. 2007. V. 292(3). P. C1070–C1077.
Hund T.J., Decker K.F., Kanter E., Mohler P.J., Boyden P.A., Schuessler R.B., Yamada K.A., Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: insights from mathematical modeling // J. Mol. Cell Cardiol. 2008. V. 45(3). P. 420–428.
Köhler A.C., Sag C.M., Maier L.S. Reactive oxygen species and excitation-contraction coupling in the context of cardiac pathology // J. Mol. Cell Cardiol. 2014. V. 73. P. 92–102.
Lehnart S.E., Maier L.S., Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart // Heart Fail. Rev. 2009. V.14 (4). P. 213–224.
Lou Q., Fedorov V.V., Glukhov A.V., Moazami N., Fast V.G., Efimov I.R. Transmural heterogeneity and remodeling of ventricular excitation-contraction coupling in human heart failure // Circulation. 2011. V. 123. P. 1881–1890.
Misra M.K., Sarwat M., Bhakuni P., Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes // Med. Sci. Monit. 2009. V. 15(10). P. RA209–RA219.
Nawata T., Takahashi N., Ooie T., Kaneda K., Saikawa T., Sakata T. Cardioprotection by streptozotocin_induced diabetes and insulin against ischemia/reperfusion injury in rats // J. Cardiovasc. Pharmacol. 2002. V. 40(4). P. 491–500.
Netticadan T., Temsah R.M., Kent A., Elimban V., Dhalla N.S. Depressed levels of Ca2+-cycling proteins may underlie sarcoplasmic reticulum dysfunction in the diabetic heart // Diabetes. 2001. V. 50(9). P. 2133–2138.
Ravingerová T., Adameová A., Matejíková J., Kelly T., Nemčeková M., Kucharská J., Pecháňová O., Lazou A. Subcellular mechanisms of adaptation in the diabetic myocardium: Relevance to ischemic preconditioning in the nondiseased heart // Exp. Clin. Cardiol. 2010. V. 15(4). P. 68–76.
Satoh N., Sato T., Shimada M., Yamada K., Kitada Y. Lusitropic effect of MCC-135 is associated with improvement of sarcoplasmic reticulum function in ventricular muscles of rats with diabetic cardiomyopathy // J. Pharmacol. Exp. Ther. 2001. V. 298(3). P. 1161–1166.
Tsutsui H., Kinugawa S., Matsushima S. Oxidative stress and heart failure // Am. J. Physiol. Heart Circ. Physiol. 2011. V. 301(6). P. H2181–H2190.
Vassallo D.V., Lima E.Q., Campagnaro P. Faria A.N., Mill J.G. Mechanisms underlying the genesis of post-extrasystolic potentiation in rat cardiac muscle // Braz. J. Med. Biol. Res. 1995. V. 28(3). P. 377–383.
Waczulikova I., Ziegelhoffer A., Orszaghova Z., Carsky J. Fluidising effect of resorcylidene aminoguanidine on sarcolemmal membranes in streptozotocin_diabetic rats: blunted adaptation of diabetic myocardium to Ca2+ overload // J. Physiol. Pharmacol. 2002. V. 53(Pt 2). P. 727–739.
Wu D., Gong C.X., Meng X., Yang Q.L. Correlation between blood glucose fluctuations and activation of oxidative stress in type 1 diabetic children during the acute metabolic disturbance period // Chin. Med. J. (Engl.). 2013. V. 126(21). P. 4019–4122.
Zhong Y., Ahmed S., Grupp I.L., Matlib M.A. Altered SR protein expression associated with contractile dysfunction in diabetic rat hearts // Am. J. Physiol. Heart Circ. Physiol. 2001. V. 281(3). P. H1137–H1147.
Ziegelhöffer A., Waczulíková I., Šikurová L., Mujkošová J., Ravingerová T. Involvement of membrane fluidity in endogenous protective processes running on subcellular membrane systems of the rat heart // Physiol. Res. 2012. V. 61 (Suppl 2). P. S11–S21.
Ziegelhöffer-Mihalovicová B., Waczulíková I., Sikurová L., Styk J., Cársky J., Ziegelhöffer A. Remodelling of the sarcolemma in diabetic rat hearts: the role of membrane fluidity // Mol. Cell Biochem. 2003. V. 249(1–2). P. 175–182.
Zima A.V., Kocksk¨amper J., Blatter L.A. Cytosolic energy reserves determine the effect of glycolytic sugar phosphates on sarcoplasmic reticulum Ca2+ release in cat ventricular myocytes // J. Physiol. 2006. V. 577(1). P. 281–293.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия биологическая