

Известия РАН. Серия физическая, 2019, T. 83, № 6, стр. 794-798
Расчеты электронной энергетической структуры Cu2Fe0.5Zn0.5SnS4 в упорядоченных структурах с различным размещением атомов Cu, Fe, Zn
Б. В. Габрельян 1, *, А. А. Лаврентьев 1, О. Ю. Хижун 2
1 Федеральное государственное бюджетное учреждение высшего образования
“Донской государственный технический университет”
Ростов на Дону, Россия
2 Институт проблем материаловедения Национальной академии наук Украины
Киев, Украина
* E-mail: boris.gabrelian@gmail.com
Поступила в редакцию 20.11.2018
После доработки 16.12.2018
Принята к публикации 25.02.2019
Аннотация
Проведены расчеты электронной энергетической структуры Cu2Fe0.5Zn0.5SnS4 в четырех моделях с упорядоченным размещением катионов в кристаллических решетках с пространственными группами I‑42m, I‑4, P‑42c, P2. Расчеты выполнены с использованием различных обменно-корреляционных потенциалов: GGA, GGA + U, модифицированного потенциала Беке–Джонсона (mBJ), c учетом антиферромагнитного упорядочения моментов атомов Fe. Выявлены парциальные состояния, формирующие валентную полосу и дно полосы проводимости соединения. Установлено, что наиболее энергетически выгодной является структура станнита, пространственная группа I‑42m.
ВВЕДЕНИЕ
При продолжающемся общем доминировании кремниевых технологий в области неорганических фотовольтаических устройств, ситуация с тонкопленочными материалами другая. Второе поколение тонкопленочных технологий базируется на бинарных (CdTe) и тройных (CuInSe2, CuInS2, CuGaS2, CuGaSe2) соединениях и их твердых растворах – Cu(In,Ga)(S,Se)2. Эффективность солнечных батарей, построенных с использованием этих технологий, достигает 20%, при том что экономически выгодными считаются ячейки с эффективностью >10%. Недостатки бинарных и тройных халькогенидов меди, в том числе относительная редкость In и Ga с учетом их широкого использования в других технологиях (например, “теплые” окна используют покрытие из In2O3), вызывает стремление найти другие соединения с подобными свойствами (подходящим значением оптической запрещенной полосы, высоким коэффициентом оптического поглощения, относительно слабой чувствительностью к дефектам), но состоящие из достаточно часто встречающихся и нетоксичных элементов. В последнее десятилетие большой интерес вызывают четырехкомпонентные халькогениды меди семейства ${\text{A}}_{2}^{{\text{I}}}$BIICIV${\text{X}}_{4}^{{{\text{VI}}}}.$ Наибольший интерес вызвали соединения Cu2ZnSnS4 и Cu2ZnSnSe4 ввиду того, что они имеют почти оптимальное значение прямой запрещенной полосы, высокий коэффициент поглощения и состоят из часто встречающихся в природе компонентов, плюс к тому имеют теоретический предел эффективности преобразования 32.2% [1]. Для получения высокого значения коэффициента преобразования солнечной энергии желательно производить чистый Cu2ZnSnS4, но на самом деле при его приготовлении возникают дополнительные бинарные и тройные фазы. Более того, Cu2ZnSnS4 содержит дефекты, вызванные обменами атомов Cu и Zn из-за их близких атомных размеров. Флуктуации электростатического потенциала Cu и Zn на дефектах CuZn + ZnCu ухудшают производительность Cu2ZnSnS4 [1]. Поиски альтернативы Cu2ZnSnS4 заставили обратить внимание в частности на соединение Cu2FeSnS4. Это полупроводник p-типа с шириной запрещенной полосы 1.28–1.5 эВ, коэффициентом поглощения, превышающим 104 cм–1 подобно Cu2ZnSnS4. На сегодняшний момент достигнута эффективность фотопреобразования ~8% [1]. В отличие от Cu2ZnSnS4, для Cu2FeSnS4 необходимо учитывать упорядочение магнитных моментов слоев, содержащих атомы железа. Согласно [2] Cu2FeSnS4, во-первых, претерпевает антиферромагнитный переход при 7 K, т.е. магнитная структура определяется ферромагнитными слоями, соединенными друг с другом антиферромагнитным образом, во-вторых, внутреннее магнитное поле на узле железа лежит в плоскости xy и, в-третьих, поле на узле олова отлично от нуля. Собственно, минерал станнит, давший название соответствующей кристаллической структуре, в идеале описывается формулой Cu2FeSnS4, а минерал кестерит – формулой Cu2ZnSnS4. Но в природе минерал станнит содержит достаточно большое количество цинка, а кестерит железа. Согласно [3] формула природного кестерита Cu1.98(Zn0.73Fe0.29 Cd0.01)Sn0.99S4.0, станнита Cu1.99(Fe0.81Zn0.18Cd0.02) Sn1.0S4.0. Поэтому актуальным является вопрос изучения свойств твердых растворов Cu2FexZn1 – xS4, в которых при некоторой концентрации x должен происходить переход от структуры станнита к структуре кестерита. Действительно, связь между станнитом и кестеритом изучалась разными авторами (см. ссылки в [4]). Одни авторы сообщали о существовании области несмешиваемости, другие, напротив, – о существовании непрерывного твердого раствора. В конце концов, были получены растворы во всей области концентраций. Было высказано предположение о переходе от структуры станнита к структуре кестерита при избытке цинка в области концентраций 60–70% [4].
КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА
Кристаллические структуры рассматриваемых бинарных, тройных и четырехкомпонентных соединений можно вывести из кубической структуры алмаза. Для бинарных соединений последовательным упорядоченным замещением одного типа атомов (алмаз) на два (CdTe), дающим в результате структуру сфалерита, в которой атомы металла (Cd) занимают свою гранецентрированную подрешетку, а атомы неметалла (Te) другую гранецентрированную подрешетку, сдвинутую относительно первой вдоль главной диагонали на четверть ее длины. Структуру тройных соединений можно получить за счет удвоения элементарной ячейки сфалерита вдоль оси z и дальнейшего замещения атомов металла одного типа (Cd) на два типа (например, Cu и In) таким образом, чтобы каждый атом неметалла был окружен двумя атомами металла одного типа (Cu) и двумя атомами металла другого типа (In), расположенными в вершинах (искаженного за счет разной длины связи разных металлов с неметаллом) тетраэдра. Получается тетраэдрическая структура халькопирита. Кристаллическая структура четырехкомпонентных халькогенидов меди может быть выведена из структуры халькопирита, подобно тому, как сама структура халькопирита выводится из структуры сфалерита. Например, заменяя два In3+ в элементарной ячейке CuInS2 (которая содержит две формульные единицы, т.е. может быть представлена формулой Cu2In2S4), на один атом Zn2+ и один атом Sn4+, можно получить изоэлектронное соединение с формулой Cu2ZnSnS4. Известно, что наиболее общими структурами для халькогенидов, основанных на меди, подобных Cu2ZnSnS4 и Cu2FeSnS4 являются тетрагональные структуры кестерита и станнита. При этом Cu2ZnSnS4 кристаллизуется в структуре кестерита (пространственная группа I-4), а Cu2FeSnS4 кристаллизуется в структуре станнита (пространственная группа I-42m), которая остается стабильной в диапазоне температур 420–500°С. При высоких температурах наблюдаются полиморфные кубические модификации с неупорядоченными сфалеритоподобными структурами. Известно, что естественные образцы Cu2FeSnS4 кристаллизуются в структуре станнита, но существуют синтезированные низкотемпературные фазы со структурами P-43m, I-4, P-4 [5]. Кроме того, в работе [6] для Cu2ZnSnS4 рассмотрены структуры, производные от структуры кестерита, получаемые при обмене атомов металлов в разных слоях, перпендикулярных оси z: P-42c – обмен атомов Cu и Zn в слое z = 1/4, P‑421c – Cu и Zn меняются местами между двумя слоями, так что образуется слой из Zn при z = 1/4 и слой из Cu при z = 3/4, P2 – атом Cu из слоя z = 1/2 меняется местами с атомом Zn из слоя z = = 3/4, что дает структуру станнита в слоях z = 1/4 и z = 3/4, но оставляет структуру кестерита в слоях z = 0 и z = 1/4.
В настоящей работе кристаллические структуры 50% растворов Fe и Zn моделировались в упорядоченных структурах I-42m, I-4, P-42c, P2 следующим образом. Тетрагональная решетка станнита (кестерита, P-42c, P2) удваивалась вдоль оси y, затем в слоях, содержащих атомы железа, половина атомов регулярным образом замещалась атомами цинка так, чтобы в ближайших слоях с этими атомами, перпендикулярных плоскости xy, были атомы железа (рис. 1). Последнее требование позволяет моделировать антиферромагнитное упорядочение магнитных моментов железа в растворе. В случае структуры P-421 c этого добиться в выбранной модели невозможно, поэтому она не рассматривалась в данной работе.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
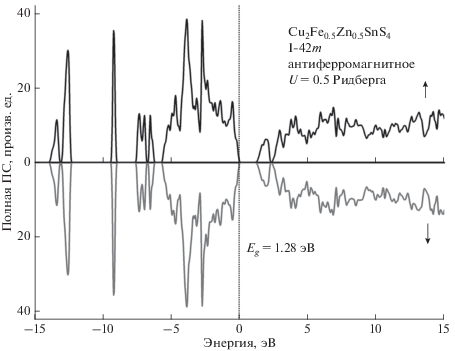
Расчеты электронной структуры проводились в приближении присоединенных плоских волн с использованием локализованных орбиталей для полуостовных состояний. Обменный потенциал строился в обобщенном градиентном приближении (GGA) по схеме PBE [7] с использованием различных поправок: GGA + U, позволяющей учитывать сильную локализацию d-электронов [8] и TB-mBJ [9], которая обычно позволяет корректировать рассчитанное значение ширины запрещенной зоны соединения. Использовался пакет программ WIEN2k [10], в котором указанное приближение и обменные потенциалы реализованы в рамках теории функционала электронной плотности. На рис. 2. приведены рассчитанные полные плотности электронных состояний Cu2ZnSnS4 в структуре, производной от структуры станнита, полученные для двух разных направлений спина. Нуль на шкале энергий соответствует положению вершины валентной полосы. При использовании обменно-корреляционного потенциала в приближении GGA плотности для состояний со спином вверх демонстрируют наличие щели между занятыми и свободными состояниями, которая исчезает для состояний со спином вниз. Это не соответствует имеющимся экспериментальным данным. Учет антиферромагнитного упорядочения магнитных моментов в слоях, содержащих атомы железа приводит к тому, что кривые плотностей состояний с разными направлениями спина практически совпадают по форме, но ширина запрещенной зоны оказывается равной нулю. Хаббардовская поправка U = 0.5 Ридберга в потенциале d-электронов железа (в нашем расчете также и меди, и цинка) “расталкивает” занятые и свободные d-состояния, в результате чего образуется запрещенная полоса с Eg = 1.28 эВ (рис. 2). Подобным же образом ведут себя и плотности состояний, рассчитанные для других структур, производных от структуры кестерита, P-42c и P2. Поэтому на рис. 3 показаны локальные парциальные состояния только для структуры, производной от структуры станнита. На рисунке видно, что вершина валентной полосы образована в основном p-состояниями серы и d-состояниями меди. В области от 2 до 5 эВ ниже вершины валентной полосы в основном представлены p-состояния серы, d-состояния меди, d-состояния железа и p-состояния олова. d-полосы железа и s‑полоса олова расположены в районе от –6 до ‒8 эВ, d-полоса цинка около –9 эВ, а s-полосы серы доминируют в области от –12 до –14 эВ. Дно полосы проводимости образовано в основном s-состояниями олова и p-состояниями серы.
Рис. 2.
Полная плотность состояний электронов с разными направлениями спина Cu2Fe0.5Zn0.5SnS4 для сверхячейки структуры станнита, рассчитанные с учетом антиферромагнитного упорядочения (afm) магнитных моментов Fe и добавки U = 0.5 Ридберга в потенциале для атомов Fe.

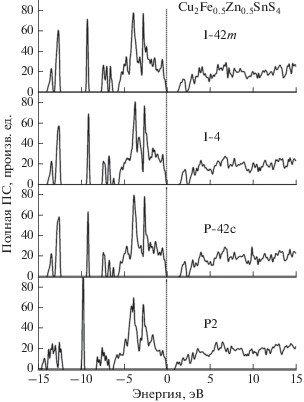
Рассчитанные в приближении GGA с учетом антиферромагнитного упорядочения магнитных моментов атомов железа в соседних слоях с поправкой GGA + U полные плотности состояний для всех четырех рассмотренных кристаллографических модификаций Cu2Fe0.5Zn0.5SnS4 приведены на рис. 4. Во всех расчетах получено полупроводниковое состояние. Ширины запрещенных полос и полные энергии электронной подсистемы для всех рассмотренных структур приведены в табл. 1.
ВЫВОДЫ
1. Наименьшее значение полной энергии получено для структуры, производной от структуры станнита, что соответствует предположениям, полученным из анализа экспериментальных данных.
2. Полная энергия структуры, производной от структуры кестерита, меньше, чем у структуры, производной от структуры P-42c, всего примерно на 2 милиридберга, что говорит о возможности сосуществования соответствующих структрных фаз.
3. В рамках использованной модели наименее энергетически выгодной является структура производная от структуры P2.
Список литературы
Vanalakar S.A., Patil P.S., Kim J.H. // Sol. Energy Mater. Sol. Cells. 2018. V. 182. P. 204.
Ganiel U., Hermon E., Shtrikman S. // J. Phys. Chem. Solids. 1972. V. 33. P. 1873.
Hall S.R., Szymanski J.T., Stewart J.M. // Can. Mineral. 1978. V. 16. P. 131.
Bonazzi P., Bindi L., Bernardini G.P., Menchetti S. // Can. Mineral. 2003. V. 41. P. 639.
Rincyn C., Quintero M., Moreno E. et al. // Sol. St. Comm. 2011. V. 151. P. 947.
Paier J., Asahi R., Nagoya A., Kresse G. // Phys. Rev. B. 2009. V. 79. Art. № 115126.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Anisimov V.I., Solovyev I.V., Korotin M.A. et al. // Phys. Rev. B. 1993. V. 48. Art. № 16929.
Tran F., Blaha P. // Phys. Rev. Lett. 2009. V. 102. Art. № 226401.
Blaha P., Schwarz K., Madsen G.K.H. et al. WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calcul. Crystal Prop. Wien: Karlheinz Schwarz. Techn. Univ., 2018.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая