

Известия РАН. Серия физическая, 2022, T. 86, № 10, стр. 1452-1456
Влияние фосфат-ионов на дианион-анионное равновесие флуоресцеина в возбужденном состоянии
Д. П. Суржикова 1, *, М. А. Герасимова 1, Е. А. Слюсарева 1
1 Федеральное государственное автономное образовательное учреждение высшего образования
“Сибирский федеральный университет”
Красноярск, Россия
* E-mail: darya19578@yandex.ru
Поступила в редакцию 01.06.2022
После доработки 15.06.2022
Принята к публикации 22.06.2022
- EDN: EILOCC
- DOI: 10.31857/S0367676522100210
Аннотация
Построены калибровочные кривые для нахождения константы равновесия флуоресцеина в возбужденном состоянии путем разложения спектра испускания на контуры дианиона и аниона. Показано, что процесс фотопереноса протона на дианион-анионной ступени диссоциации конкурирует с излучательными процессами, причем его эффективность растет с увеличением концентрации фосфат-ионов, выступающих в роли акцепторов протонов. Изменение кажущейся константы диссоциации $\Delta {\text{p}}{{K}_{{app2}}}$ при фотовозбуждении в полосу поглощения дианиона (488 нм) близко к предельному значению, найденному по циклу Фёрстера (ΔpKa = −0.71).
ВВЕДЕНИЕ
Сигнал большинства оптических pH-индикаторов основан на изменении спектральных свойств в зависимости от зарядового состояния флуорофора (например, при протонировании). Чувствительность таких индикаторов определяется углом наклона линейного участка сигмоидной зависимости концентрации соответствующей ионной формы от pH, следующей из уравнения Хендерсона–Хассельбаха [1]. Однако этот метод определения pH имеет ограниченную применимость в реальных биологических системах из-за того, что ионные равновесия в основном состоянии могут быть сильно смещены вследствие высокой концентрации неорганических ионов. Так, при увеличении ионной силы до 3 М изменение константы диссоциации pKa в основном состоянии, определенное оптическими методами, может составлять более 1.2 единиц рН [2].
Равновесие возбужденного состояния зачастую смещено относительно основного состояния, благодаря чему индикатор становится фотокислотой или фотооснованием. Изменение pKa при фотовозбуждении в рамках термодинамического цикла Фёрстера [3] пропорционально разности частот 0–0 переходов депротонированной (А−) и протонированной (AH) форм:
(1)
$\Delta {\text{p}}{{K}_{a}} = h\frac{{\nu _{{00}}^{{{{{\text{A}}}^{ - }}}} - \nu _{{00}}^{{{\text{AH}}}}}}{{2.3kT}}.$Ионное равновесие в возбужденном состоянии также подвержено влиянию неорганических ионов, присутствующих в растворе. Из-за конкуренции между переносом протона и испусканием фотона, флуоресцентный сигнал становится весьма чувствительным к концентрации доноров/акцепторов протонов.
Флуоресцеин является примером pH-индикатора, дианион-анионное равновесие которого приходится на физиологический диапазон pH (pKa 6.31 [4]), что обуславливает его привлекательность для использования в качестве внутриклеточного pH зонда [5]. Было обнаружено, что на этой ступени диссоциации флуоресцеин является фотокислотой [6–10], а опубликованные значения константы диссоциации в возбужденном состоянии ${\text{p}}K_{a}^{*}$ находятся в пределах 5.54–6.31 [6, 9, 10]. Подобный разброс значений связан, как правило, с различием типов и концентрацией окружающих ионов, а также с методикой определения самой константы диссоциации.
В настоящей работе мы оценили влияние фосфат-ионов (равновесная смесь моно- и дигидрофосфат-ионов буферного раствора) на ионное равновесие флуоресцеина в основном и возбужденном состояниях. Для этого мы впервые использовали разложение наблюдаемого контура поглощения и испускания при разном pH на контуры ионных форм с определением их амплитудного вклада. Фосфат-ионы стимулируют реакцию фотопереноса протона, участвуя в регулировании метаболизма и в поддержании гомеостаза в реальных биологических системах. Понимание степени влияния фосфат-ионов на дианион-анионное равновесие в возбужденном состоянии способствует расширению сенсорных приложений флуоресцеина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы
В работе была использована динатриевая соль флуоресцеина С20H10Na2O5 (Fluka). В качестве растворителя использовался фосфатный буфер (pH 5.6–7.1) при трех значениях суммарной молярности фосфат-ионов (${{{\text{H}}}_{2}}{\text{PO}}_{4}^{ - }$, ${\text{HPO}}_{4}^{{2 - }}$) [CP]: 0.1, 0.5 и 1 М. Для приготовления растворов с pH, выходящим за пределы указанного диапазона, использовались соляная кислота HCl и гидроксид калия KOH. В исследованном диапазоне pH флуоресцеин находится преимущественно в двух ионных формах: дианионной (диссоциирован по карбоксильной и гидроксильной группам) и анионной (диссоциирован по карбоксильной группе); в меньшей степени в цвиттерионной нейтриальной форме (константа равновесия анион-нейтрального равновесия 3.46 [4]). Концентрация красителя в растворах составляла около 8 ⋅ 10−6 М. Измерения проводились через 30 мин после приготовления раствора.
Контроль pH раствора осуществлялся с помощью pH-метра SevenCompact S220 (Mettler Toledo, Швейцария). Спектры поглощения были получены на спектрофотометре Lambda 35 (PerkinElmer, США). Спектры испускания измерялись на спектрофлуориметре Fluorolog 3-22 (Horiba Jobin Yvon, США) при при возбуждении в полосе поглощения аниона (λexc = 435 нм) и дианиона (λexc = 488 нм). Полученные спектры корректировались на спектральную чувствительность детектора и реабсорбцию. Все измерения проводились при комнатной температуре с использованием кюветы сечением 1 × 1 см при L-геометрии возбуждения.
Разложение составных спектров
Для разложения экспериментальных спектров поглощения и испускания на составляющие, соответствующие отдельным ионным формам, использовалось программное обеспечение OriginPro (OriginLab, США), реализующее алгоритм нелинейной аппроксимации Левенберга–Марквардта. Качество разложения оценивалось с использованием взвешенных остатков и значения χ2. Использовались контуры поглощения дианионной (D), анионной (M) и цвиттерионной (Z) форм [4], а также контуры испускания M* и D* форм [11].
Определение констант диссоциации в основном и возбужденном состояниях
Концентрации ионных форм флуоресцеина в основном состоянии были рассчитаны на основании полученных амплитудных вкладов каждой формы в измеренные спектры поглощения с учетом известных молярных коэффициентов экстинкции в максимуме спектров (87 700 М–1 ⋅ см–1 для D, 32 700 М–1 ⋅ см–1 для М, 55 000 М–1 ⋅ см–1 для Z [4]). Полученное значение для дианионной формы D нормировалось на его максимальное значение при pH 10. Зависимость от pH аппроксимировалась функцией Больцмана. Значение кажущейся (полученной без контроля ионной силы раствора) константы ${\text{p}}{{K}_{{app}}}$ определялось как середина перехода в pH шкале.
Аналогичным образом, на основании полученных амплитудных вкладов каждой формы в измеренные спектры испускания с учетом известных квантовых выходов (0.93 для D и 0.36 для М [4]) были рассчитаны концентрации ионных форм флуоресцеина в возбужденном состоянии. Полученное значение для дианионной формы D нормировалось на его максимальное значение при pH 10. Зависимость от pH аппроксимировалось функцией Больцмана. Кажущаяся константа диссоциации в возбужденном состоянии ${\text{p}}K_{{app}}^{*}$ определялась как значение середины перехода.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ионное равновесие в основном состоянии
Калибровочные кривые для нахождения константы диссоциации в основном состоянии представлены на рис. 1. Найденное значение ${\text{p}}{{K}_{{app}}}$ при концентрации фосфат-ионов 1 M близко к опубликованному ранее [9]. Присутствие фосфат-ионов незначительно смещает дианион-анионное равновесие в сторону больших значений pH (табл. 1, рис. 1). Это смещение (около 0.1 ед. pH), тем не менее, оказывает влияние на концентрацию ионных форм в основном состоянии. Так, при pH 6.3 разница концентраций D формы при концентрации фосфат-ионов 0.1 и 1 M достигает 20%. Влияние ионов различной химической природы на смещение константы диссоциации достаточно специфично. Так, при увеличении концентрации KCl до ∼0.7 M дианион-анионное равновесие флуоресцеина смещается в сторону меньших значений pH, а при дальнейшем увеличении растет [12].
Рис. 1.
Калибровочная кривая для нахождения константы дианион-анионной диссоциации ${\text{p}}K_{{app}}^{{}}$ в основном состоянии. Вставка – пример разложения спектра поглощения на составляющие, из которого была найдена нормированная концентрация дианиона.
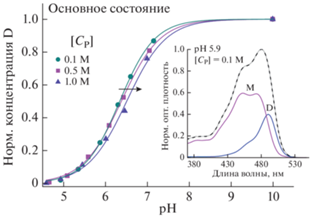
Таблица 1.
Константы диссоциации флуоресцеина в основном и возбужденном состояниях
| [CP], M | ${\text{p}}{{K}_{{app}}}$ | ${\text{p}}K_{{app1}}^{*}$ | $\Delta {\text{p}}K_{{app{\text{1}}}}^{{}}$ | ${\text{p}}K_{{app{\text{2}}}}^{*}$ | $\Delta {\text{p}}K_{{app{\text{2}}}}^{{}}$ |
|---|---|---|---|---|---|
| λexc = 435 нм | λexc = 488 нм | ||||
| 0.1 | 6.35 ± 0.03 | 6.34 ± 0.04 | –0.01 ± 0.05 | 5.42 ± 0.04 | –0.93 ± 0.07 |
| 0.5 | 6.39 ± 0.03 | 5.99 ± 0.03 | –0.40 ± 0.04 | 5.44 ± 0.04 | –0.95 ± 0.08 |
| 1 | 6.52 ± 0.04 | 5.92 ± 0.06 | –0.60 ± 0.06 | 5.62 ± 0.04 | –0.90 ± 0.09 |
Ионное равновесие в возбужденном состоянии
Рассчитанное по (1) значение разницы констант диссоциации в возбужденном и основном состоянии флуоресцеина $\Delta {\text{p}}{{K}_{a}} = - 0.71$ единиц pH. Оно соответствует предельной ситуации, при которой за время жизни возбужденного состояния успевает полностью установиться новое ионное равновесие. Однако этого может не произойти, если излучательные (или другие релаксационные процессы) окажутся более вероятными. В этом случае краситель перейдет в основное состояние прежде, чем изменит свое ионное состояние, и кажущаяся константа диссоциации $\Delta {\text{p}}{{K}_{{app}}}$ окажется по модулю меньше ожидаемого значения. При вероятности установления равновесия ниже вероятности процессов дезактивации возбуждения $\Delta {\text{p}}{{K}_{{app}}}$ может быть близко к нулю. Промежуточное значение говорит от том, что обсуждаемые процессы сравнимы по скорости.
Кажущаяся константа диссоциации $\Delta {\text{p}}{{K}_{{app}}}$ в возбужденном состоянии проявляет сильную зависимость от λexc (рис. 2 и 3, табл. 1). Для [CP] = = 0.1 M разница между данными, полученными при двух длинах волн возбуждения, составила почти единицу pH. При pH 6.3 при возбуждении на длине волны 435 нм разница концентраций D формы в присутствии 0.1 и 1 M фосфат-ионов составляет 35%, а при возбуждении на длине волны 488 нм – 11%. Зависимость концентрации возбужденного состояния от длины волны возбуждения указывает на то, что за время жизни возбужденного состояния ионное равновесие не успевает установиться.
Рис. 2.
Калибровочная кривая для нахождения константы дианион-анионной диссоциации ${\text{p}}K_{{app{\text{1}}}}^{*}$ в возбужденном состоянии. Вставка – пример разложения спектра испускания (возбуждение 435 нм) на составляющие, из которого была найдена нормированная концентрация дианиона.

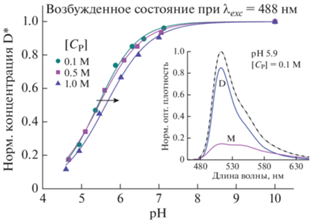
Рис. 3.
Калибровочная кривая для нахождения константы дианион-анионной диссоциации ${\text{p}}K_{{app{\text{2}}}}^{*}$ в возбужденном состоянии. Вставка – пример разложения спектра испускания (возбуждение 488 нм) на составляющие, из которого была найдена нормированная концентрация дианиона.
При обсуждении зависимости ${\text{p}}K_{{app{\text{1}}}}^{*}$ от концентрации фосфат-ионов при λexc = 435 нм (табл. 1) примем во внимание, что концентрации H+ и OH− низки в исследуемой области рН 5–8, поэтому молекулы воды не акцептируют протоны достаточно эффективно, чтобы конкурировать с процессами релаксации возбужденного состояния [9]. Реакцию депротонирования аниона можно ускорить путем увеличения концентрации фосфат-ионов. В исследуемой области pH основной протон-акцепторной формой являются ионы ${\text{HPO}}_{4}^{{2 - }}$. В [9] было показано, что при низких концентрациях фосфат-ионов, перенос протона не происходит. Согласно нашим данным, фосфат-ионы при концентрации более 0.1 М запускают эту реакцию ($\Delta {\text{p}}{{K}_{{app1}}} \ne 0$). Грубая экстраполяция зависимости $\Delta {\text{p}}{{K}_{{app1}}}$ от концентрации фосфат-ионов к предельному значению, полученному при возбуждении на 488 нм, показывает, что при концентрации фосфат-ионов около 2.5–3 М достигается ионное равновесие.
При длине волны возбуждения 488 нм значение $\Delta {\text{p}}K_{{app{\text{2}}}}^{{}}$ близко предельному значению (−0.71), что говорит о том, что скорость релаксации возбуждения дианиона меньше, чем у аниона. Близкое значение $\Delta {\text{p}}{{K}_{a}} = - 0.64$ было получено в [10] методом время-разрешенной флуоресцентной спектроскопии.
Рис. 4.
$\Delta {\text{p}}{{K}_{{app{\text{1}}}}}$и $\Delta {\text{p}}{{K}_{{app{\text{2}}}}}$ при длинах волн возбуждения 435 и 488 нм при вариации концентрации фосфат-ионов. Пунктиром нанесено предельное значение $\Delta {\text{p}}{{K}_{a}} = - 0.71$, рассчитанное по (1). Добавлена линия предполагаемой зависимости $\Delta {\text{p}}K_{{app{\text{1}}}}^{{}}$ до достижения предельного значения.

ЗАКЛЮЧЕНИЕ
Анализ вклада контуров отдельных ионных форм в регистрируемые спектры поглощения и испускания дает возможность построения калибровочных кривых для расчета констант диссоциации в основном и возбужденном состояниях. Длина волны возбуждения и концентрация фосфат-ионов в целом регулируют протекание реакций, определяющих соотношение анионов и дианионов флуоресцеина в возбужденном состоянии, то есть, ионное равновесие в условиях возбуждения. Константы диссоциации в возбужденном состоянии при преимущественном возбуждении анионов оказались зависящими от концентрации (моно)гидрофосфат-ионов, выступающих в изучаемой системе в качестве акцепторов протонов в реакции фотопереноса протона. Увеличением концентрации фосфат-ионов более 2.5 М можно повысить эффективность протекания реакции фотопереноса и достичь дианион-анионного равновесия за время жизни возбужденного состояния.
Работа выполнена при поддержке Российского научного фонда (проект № 22-22-00724). Авторы благодарят А. Плотникова за помощь в анализе экспериментальных данных.
Список литературы
Po H.N., Senozan N.M. // J. Chem. Educ. 2001. V. 78. No. 11. P. 1499.
Edmonds T.E., Flatters N.J., Jones C.F. et al. // Talanta. 1988. V. 35. P. 103.
Valeur B., Berberan-Santos M.N. Molecular fluorescence: principles and applications. Weinheim: Wiley-VCH, 2012.
Klonis N., Sawyer W.H. // J. Fluoresc. 1996. V. 6. No. 3. P. 147.
Le Guern F., Mussard V., Gaucher A. et al. // Int. J. Mol. Sci. 2020. V. 21. No. 23. Art. No. 9217.
Gerasimova M.A., Tomilin F.N., Malyar E.Yu. et al. // Dyes Pigm. 2020. V. 173. Art. No. 107851.
Alvarez-Pez J.M., Ballesteros L., Talavera E. et al. // J. Phys. Chem. A. 2001. V. 105. P. 6320.
Surzhikova D., Gerasimova M., Tretyakova V. et al. // J. Photochem. Photobiol. A. 2021. V. 413. Art. No. 113233.
Yguerabide J., Talavera E., Alvarez J.M. et al. // Photochem. Photobiol. 1994. V. 60. P. 435.
Слюсарева Е.А., Герасимова М.А. // Изв. вузов. Физ. 2013. Т. 56. № 12. С. 48; Slyusareva E.A., Gerasimova M.A. // Russ. Phys. J. 2014. V. 56. No. 12. P. 1370.
Суржикова Д.П., Герасимова М.А., Слюсарева Е.А. // Изв. вузов. Физ. 2021. Т. 64. № 11. С. 108; Surzhikova D.P., Gerasimova M.A., Slyusareva E.A. // Russ. Phys. J. 2022. V. 64. No. 11. P. 2102.
Мчедлов-Петросян Н.О. // Вест. Харьков. нац. ун-та. 2004. Т. 626. № 11(34). С. 221.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая