

Химическая физика, 2020, T. 39, № 9, стр. 9-17
Квантовохимическое моделирование адсорбции водорода на кластерах AunNim и AunCum, n + m = 13
Н. В. Дохликова 1, *, А. К. Гатин 1, С. Ю. Сарвадий 1, Е. И. Руденко 1, М. В. Гришин 1, Б. Р. Шуб 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: dohlikovanv@chph.ras.ru
Поступила в редакцию 22.05.2019
После доработки 25.09.2019
Принята к публикации 21.10.2019
Аннотация
В работе представлены результаты квантовохимического моделирования адсорбции водорода на биметаллических наночастицах золото–никель и золото–медь. Установлено, что изменения адсорбционных свойств биметаллической наносистемы определяются изменением расстояний между атомами и перераспределением электронной плотности в объеме кластера, что приводит к увеличению энергии связи атома водорода в окрестности атомов Ni/Cu и уменьшению – в окрестности атомов Au кластера биметалла.
ВВЕДЕНИЕ
Исследования биметаллических наночастиц в настоящее время получили широкое распространение в нанокатализе, благодаря уникальным физико-химическим свойствам наноструктур этого типа, таким как высокая селективность [1], реакционная способность [2] и высокая стабильность [3]. Но наибольший интерес вызывает сильное изменение свойств биметаллических наносистем по сравнению со свойствами монометаллических наносистем того же элементного состава. Происхождение этого эффекта связано с взаимодействием между различными атомами биметалла, а также с деформацией геометрической структуры за счет различных постоянных решетки кристаллов исходных веществ [4]. Совместное действие этих факторов и может привести к нелинейному изменению параметров биметаллической системы, т.е. к синергетическому эффекту. В реальных биметаллических наночастицах взаимное влияние атомов разных типов друг на друга и на протекающие процессы, например, адсорбцию газов, может иметь очень запутанный характер благодаря большому количеству варьируемых параметров, что усложняет описание и исследование таких наносистем [5]. Однако в численных экспериментах можно осуществить точный контроль изменений атомной и электронной структур моделей биметаллических наночастиц и достичь таким образом лучшего понимания каталитических реакций с их участием.
В данной работе изложены результаты численного моделирования наночастиц, состоящих из атомов золота и никеля, а также золота и меди. Согласно литературным данным, варьирование элементного состава наноструктур на основе золота с добавлением атомов переходного или благородного металлов может привести к сильному изменению адсорбционных и каталитических свойств получившейся биметаллической наносистемы. Например, в работе [6] установлено, что допирование кластеров золота (AuN, N = 2–7) атомами переходных металлов группы платины Ni, Pt, Pd увеличивает стабильность кластера биметалла. При этом его адсорбционные и электронные свойства определяются особенностями взаимодействия внешних электронных орбиталей атомов золота и примеси, а именно степенью перекрытия d–d и s–d орбиталей. Атомы Ni и Pt усиливают взаимодействие между атомами в кластере благодаря большему перекрыванию s-орбиталей, по сравнению с атомами Pd. Степень s–d-гибридизации и выраженности релятивистских эффектов также влияет на тенденцию к образованию планарных структур, которая наиболее сильна в кластерах Au–Pt и Au–Pd. В работе [7] с помощью теории функционала плотности (DFT) проводилось исследование окисления СО на кластерах MAu6 (M = Ni, Pt, Pd). Расчеты показали, что при допировании кластера Ni кислород адсорбируется на кластере атомарно. В работе [8] с помощью методов DFT и микрокинетического моделирования установлено, что увеличение содержания меди в биметаллических кластерах AuCu приводит к снижению реакционного барьера для реакции окисления CO. При этом может ухудшаться селективность за счет сильного связывания с СО.
Очевидно, что биметаллические наночастицы представляют собой системы со сложной атомной и электронной структурой, которые формируется под влиянием множества факторов. Поиск ключевых факторов, влияющих на каталитические свойства биметаллических наночастиц является одной из главных целей как настоящей работы, так и нанокатализа в целом.
Ранее нами было проведено исследование трансформации атомной и электронной структур кластеров Au и Au–Ni при взаимодействии с атомарным водородом [9, 10]. Согласно полученным данным, адсорбция водорода на кластере золота вызывает локальное снижение плотности состояний вблизи уровня Ферми, что в рамках резонансной модели хемосорбции означает уменьшение химической активности кластера при том, что золото является химически инертным материалом. Допирование никелем приводит к перераспределению электронной плотности кластера, в результате которого атомы золота приобретают отрицательный заряд, а атомы никеля – положительный. Как показали расчеты, адсорбция водорода в окрестности атомов золота становится энергетически более выгодной. В настоящем исследовании дополненные результаты предыдущих работ используются для сопоставления физико-химических свойств золотых и золото-медных кластеров. Цель нашей работы – определение качественных тенденций изменения адсорбционных свойств биметаллических кластеров золото–медь и золото–никель на примерах взаимодействия с атомарным водородом при варьировании элементного состава кластера и мест адсорбции атома водорода
МЕТОДИКА ИССЛЕДОВАНИЙ
Численный эксперимент, моделирующий взаимодействие водорода с биметаллическими наночастицами золото–медь и золото–никель, проведен в рамках теории функционала плотности. Для расчетов использовались программные пакеты Quantum Espresso-5.1.1 (QE) [11] и OpenMX 3.8 (OMX) [12]. В QE использовался стандартный ультрамягкий псевдопотенциал, сгенерированный в обобщенном градиентном приближении, с функционалом PBE и учетом релятивистских поправок движения электронов, взятый из библиотеки псевдопотенциалов QE [13]. Энергия отсечки базисного набора – 24 Ry. Объем ячейки – 20 × 20 × 20 Å3. Спиновая поляризация не учитывалась.
Контрольный расчет энергии связи 13-ти атомных икосаэдрических изомеров кластеров Au13, Cu13 и Ni13 с атомом Н, адсорбированным над одним атомом кластера, в стандартных псевдопотенциалах PZ и PBE, сгенерированных в приближениях LDA и GGA соответственно, показал, что при одинаковом наборе исходных параметров численного эксперимента относительные тенденции изменения рассчитываемых величин сохраняются (табл. 1):
Таблица 1.
Энергия связи атома водорода с кластерами Au13, Cu13 и Ni13 в псевдопотенциалах PZ (LDA) и PBE (GGA)
| Кластер | Ebond (PZ), эВ | Ebond (PBE), эВ |
|---|---|---|
| Au13 | –2.90 | –2.64 |
| Cu13 | –3.08 | –2.71 |
| Ni13 | –3.37 | –3.09 |
Так как оба использованных нами псевдопотенциала качественно дают одни и те же результаты, то мы выбрали псевдопотенциал в наиболее подходящем для металлических систем приближении GGA.
В OMX описание электронной структуры кластеров проводилось с помощью стандартного базисного набора [12]: Au – s4p3d2f2, Cu – s3p3d3, Ni – s4d4d3f2, H – s3p2. Выбор радиуса обрезания радиальной части волновых функций основан на тестовых расчетах центров d-зон кластеров Au13, Cu13 и Ni13 (табл. 2). Для удобства в дальнейшем электронная структура будет описываться в терминах твердого тела. Под центром d-зоны здесь имеется в виду рассчитанный центр распределения d-орбиталей атомов кластера. Как видно из табл. 2, для Au достаточным является радиус 9 Å; для Cu – 8 Å, при радиусе 10 Å значение центра d‑зоны завышено; для Ni – 10 Å, поскольку известно, что его d-зона не заполнена; для H был взят максимально возможный в OMX радиус обрезания 7 Å для более подробного описания атома адсорбата.
Таблица 2.
Центр d-зоны кластеров Au13, Cu13 и Ni13 при различных радиусах обрезания радиальной части волновых функций базиса атомов Au, Cu, Ni
| Кластер | Радиус, Å | Центр d-зоны, эВ |
|---|---|---|
| Au13 | 7 9 11 |
–2.20 –2.26 –2.76 |
| Cu13 | 6 8 10 |
–0.85 –0.66 –0.08 |
| Ni13 | 6 8 10 |
–0.79 –0.53 0.04 |
Для краткости далее в статье положение и место адсорбции атома водорода на кластере будет называться сайтом. Положение адсорбированного атома над одним атомом кластера называется одноцентровый сайт адсорбции, над двумя – двуцентровый, над тремя – трехцентровый.
В рамках интерпретации качественных явлений, наблюдаемых в экспериментах, модель малого кластера удовлетворительно воспроизводит электронную структуру ограниченной системы атомов, которой является наночастица, и изменения, происходящие с ней при взаимодействии с другими атомами, в данном случае с водородом.
Методика исследований включает в себя расчет и сопоставление энергий связи с адсорбатом, а также локальных энергетических спектров атомов кластера с локальной проводимостью туннельного контакта, образованного зондом сканирующего туннельного микроскопа (СТМ), и наночастицей, нанесенной на поверхность графита. В экспериментах по исследованию результатов адсорбции водорода на поверхности золотых наночастиц и биметаллического наноструктурированного покрытия с применением СТМ использовались малые напряжения смещения U. При этом измерялись зависимости проводимости туннельного контакта от приложенного к нему напряжения I(U) (вольт-амперные характеристики, ВАХ). Если матричный элемент перекрытия волновых функций зонда и атома поверхности, M, аппроксимировать усредненным по области туннельного контакта значением и считать, что плотность состояний зонда, ρtip(0), не имеет особенностей в окрестности уровня Ферми, то туннельный ток (и проводимость контакта) определяется только плотностью состояний образца, ρsample(ε), где ε – энергия:
(1)
$I(U) = {\text{const}}M{{\rho }_{{tip}}}(0)\int\limits_{ - eU}^0 {{{\rho }_{{sample}}}(\varepsilon )d\varepsilon } .$Сопоставляя измеренные в СТМ после экспозиции в водороде локальные ВАХ и рассчитанные для соответствующих адсорбционных комплексов плотности состояния, можно не только объяснить результаты эксперимента, но и получить новую информацию о химических свойствах наночастиц. Как известно, кластеры металлов имеют большое количество близких по энергиям изомеров. Так как в рамках данной работы не требуются исследования атомной и электронной структур кластеров как отдельных объектов, то для однозначности выбраны 13-атомные икосаэдрические изомеры, как симметричные кластеры, обладающие полностью заполненной атомной оболочкой и являющиеся локальным минимумом среди ряда изомеров. Стоит отметить, что во избежание перехода к другому изомеру при расчете адсорбции водорода, положения атомов кластера фиксировались.
КВАНТОВОХИМИЧЕСКОЕ МОДЕЛИРОВАНИЕ
В качестве исходных моделей биметаллических наночастиц использованы кластеры типа AunMm (n + m = 13, M = Ni, Cu). Атомная структура биметаллических кластеров создавалась путем замены атомов на поверхности икосаэдрических изомеров кластеров Au13, Ni13 и Cu13 и последующей релаксации с помощью квазиньютоновских методов [14], т.е. осуществлялся поиск кластера с минимальной энергией. Полученные биметаллические кластеры соответствовали локальным минимумам энергии. Для дальнейшего исследования биметаллических кластеров были выбраны следующие:
1. имитирующие единичные дефекты на поверхности наночастицы – Au12Ni1, Ni12Au1, Au12Cu1, Cu12Au1 с примесным атомом на поверхности кластера;
2. моделирующие наночастицу с примерно одинаковым числом равномерно распределенных атомов – Au7Ni6, Au7Cu6. Ниже эти наночастицы будут называться “аморфными”;
3. янус-кластеры, у которых атомы разного типа сегрегированы и имеют границу раздела – jan-Au7Ni6, jan-Ni7Au6, jan-Au7Cu6, jan-Cu7Au6.
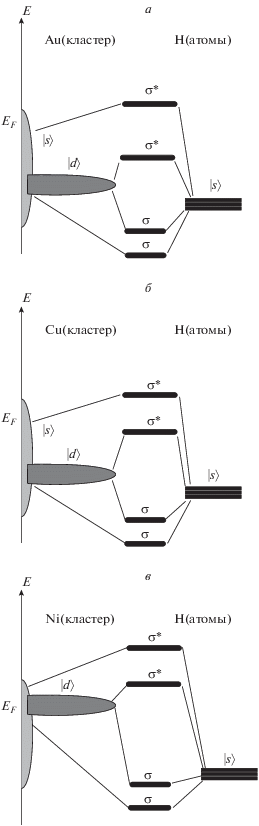
Исследование особенностей взаимодействия атома H с указанными выше кластерами проводилось путем расчета параметров связи (табл. 3) и спроектированных плотностей состояний при различных положениях адсорбированного атома H на поверхности, т.е. на различных сайтах. Вначале сделаем некоторые предположения об особенностях взаимодействия атома H с кластерами Au13, Cu13, Ni13 (рис. 1). На этом рисунке приведены кривые спроектированных плотностей состояний атомов: исходная (PDOS1) и после адсорбции Н (PDOS2), а также на врезках показаны структуры кластеров. Рисунок 2 иллюстрирует расположение молекулярных орбиталей для кластеров различного состава. В рамках модели резонансной хемосорбции ключевым фактором при адсорбции атома H на этих кластерах, определяющим здесь основные различия взаимодействия, является дефект резонанса |ɛd – ɛa| [15]. Поскольку центр d-зоны атомов Au и Cu лежит ниже уровня Ферми (рис. 1а), это приводит к появлению заполненного связывающего (σ) и заполненного антисвязывающего (σ*) состояний d-зоны Au13 и Cu13 и s-орбиталей атомов H (рис. 2а, б). При этом сдвиг центра d-зоны Cu13 немного больше, чем у Au13, что коррелирует с большей энергией связи (табл. 3). Антисвязывающее состояние σ* Au13 лежит ближе к уровню Ферми, что объясняется большим значением матричного элемента связывания |Vad| по сравнению тем же параметром для Cu13, т.е. большим вкладом отталкивательного паулиевского взаимодействия у Au [16]. Следствием этого обстоятельства также может быть то, что наиболее энергетически стабильным для кластера Au13 является двуцентровый сайт адсорбции атома H, а не трехцентровый, как у кластеров Cu13 и Ni13. Дефект резонанса для кластера Ni13 и атома H больше, т.е. центр d-зоны Ni находится в околофермиевской зоне (рис. 1в), поэтому антисвязывающее состояние σ* лежит выше уровня Ферми и не заполнено (рис. 2в), что приводит к увеличению энергии связи кластера Ni13 с атомом H (табл. 3).
Таблица 3.
Вычисленные методом DFT-моделирования параметры кластеров AunMm (n + m = 13, M = Ni, Cu) и их взаимодействия с атомом H
| Кластер | Rl, Å | Rc, Å | Rb, Å | Eb, эВ | S | dc, эВ | dc H, эВ | ∆d | qMul, e |
|---|---|---|---|---|---|---|---|---|---|
| Au13 | 2.85 | 2.73 | 1.75 | –3.20 | 2 | –2.85 | –3.27 | 0.42 | 0.0 |
| Ni13 | 2.42 | 2.32 | 1.75 | –3.33 | 3 | –0.028 | –0.045 | 0.017 | 0.0 |
| Cu13 | 2.50 | 2.35 | 1.75 | –3.29 | 3 | –0.45 | –0.97 | 0.52 | 0.0 |
| Au12Ni1 | 2.92(Au) 2.64(Ni) |
2.72(Au) 2.46(Ni) |
1.75(s1) | –3.20 | 2 | –2.80 | –3.28 | 0.48 | –0.04(Au) +0.48(Ni) |
| 1.57(s2) | –3.16 | 1 | –2.78 | –3.42 | 0.64 | ||||
| Au7Ni6 | 2.50(Au–Ni) | 2.77(Au) 2.50(Ni) |
1.61(Ni) 1.81(Au)(s1) |
–2.87 | 2 | 0.36 –2.69 |
0.24 –2.97 |
0.12 0.28 |
–0.32(Au) +0.37(Ni) |
| 1.60(s2) | –2.80 | 1 | –2.69 | –3.29 | 0.60 | ||||
| Ni12Au1 | 2.70(Au) 2.37(Ni) |
2.62(Au) 2.32(Ni) |
1.60(s1) | –3.02 | 1 | –3.02 | –3.39 | 0.37 | –0.24(Au) +0.02(Ni) |
| 1.75(s2) | –3.35 | 3 | –0.022 | –0.042 | 0.02 | ||||
| jan-Au7Ni6 | 2.78(Au) 2.32(Ni) 2.52(Au–Ni) |
нет центрального атома | 1.59(sv(Au)) | –2.53 | 1 | –3.20 | –4.03 | 0.83 | –0.13(Au) +0.15(Ni) |
| 1.59(si(Au)) | –3.15 | 1 | –2.87 | –3.50 | 0.63 | ||||
| 1.73(sv(Ni)) | –3.52 | 2 | –0.039 | –0.095 | 0.056 | ||||
| 1.73(si(Ni)) | –3.46 | 3 | –0.064 | –0.14 | 0.076 | ||||
| jan-Ni7Au6 | 2.88(Au) 2.34(Ni) 2.59(Au–Ni) |
2.59(Au) 2.39(Ni) |
1.58(sv(Au)) | –3.02 | 1 | –3.15 | –3.20 | 0.05 | –0.11(Au) +0.10(Ni) |
| 1.59(si(Au)) | –3.08 | 1 | –3.12 | –3.50 | 0.38 | ||||
| 1.74(sv(Ni)) | –3.44 | 3 | 0.089 | 0.018 | 0.071 | ||||
| 1.74(si(Ni)) | –3.44 | 3 | 0.084 | 0.012 | 0.072 | ||||
| Au12Cu1 | 2.87(Au) 2.68(Cu) |
2.72(Au) 2.49(Cu) |
1.57(s1,s2) | –3.16 | 1 | –2.83 | –3.42 | 0.59 | –0.03(Au) +0.34(Cu) |
| Au7Cu6 | 2.52(Au–Cu) | 2.92(Au) | 1.59(Au)(s1) | –3.09 | 1 | –3.20 | –3.67 | 0.47 | –0.26(Au) +0.31(Cu) |
| 1.66(Cu) |
–2.90 | 2 | –0.86 |
–0.99 |
0.13 |
||||
| 1.73(Au)(s2) | –3.27 | –3.26 | –0.01 | ||||||
| Cu12Au1 | 2.68(Au) 2.45(Cu) |
2.59(Au) 2.35(Cu) |
1.60(s1) | –3.00 | 1 | –3.16 | –3.72 | 0.56 | –0.44(Au) +0.04(Cu) |
| 1.74(s2) | –3.33 | 3 | –0.83 | –1.08 | 0.25 | ||||
| jan-Au7Cu6 | 2.98(Au) 2.58(Cu) 2.62(Au–Cu) |
2.68(Au) 2.49(Cu) |
1.57(sv(Au)) | –3.18 | 1 | –2.74 | –3.13 | 0.39 | –0.13(Au) +0.15(Cu) |
| 1.57(si(Au)) | –3.23 | 1 | –2.83 | –3.44 | 0.61 | ||||
| 1.74(sv(Cu)) | –3.37 | 3 | –0.50 | –0.84 | 0.34 | ||||
| 1.67(si(Cu)) | –3.25 | 2 | –0.52 | –0.90 | 0.38 | ||||
| jan-Cu7Au7 | 2.89(Au) 2.48(Cu) 2.58(Au–Cu) |
2.62(Au) 2.43(Cu) |
1.57(sv(Au)) | –3.00 | 1 | –2.83 | –3.20 | 0.37 | –0.13(Au) +0.11(Cu) |
| 1.58(si(Au)) | –3.16 | 1 | –2.93 | –3.61 | 0.68 | ||||
| 1.73(sv(Cu)) | –3.40 | 3 | –0.66 | –0.91 | 0.25 | ||||
| 1.73(si(Cu)) | –3.40 | 3 | –0.66 | –0.92 | 0.26 |
Примечание: Rl – среднее расстояние между поверхностными атомами, Rc – среднее расстояние до центрального атома, Eb, Rb– энергия и длина связи с атомом H; S – одно-, двух- или трехцентровое положение (сайт) адсорбированого атома H; dc, dcH – центр d-зоны для кластеров без атома H и с H, соответственно; ∆d – сдвиг d-зоны, +/– – вниз/вверх; qMul – заряд атома по Малликену. Сайты адсорбата обозначаются как s1 – на примесном атоме иного сорта, s2 – на соседнем с примесным атомом атоме, si(Au/Cu/Ni) – на интерфейсе янус-кластеров, sv(Au/Cu/Ni) – на максимальном удалении от интерфейса янус-кластеров. В скобках рядом с сайтами si и sv указывается соответствующая подсистема металла.
Рис. 1.
Изменения спроектированных плотностей состояний атомов кластеров Au13 (а), Cu13 (б) и Ni13 (в) при взаимодействии с атомом H. На всех графиках: PDOS1 – исходная спроектированная плотность состояний (1), PDOS2 – плотность состояний после адсорбции H (2); ∆d – сдвиг d-зоны (+/– – вниз/вверх), Eb – энергия связи с атомом Н, EF – энергия Ферми. На вставках приведены атомные структуры кластеров. Атомы, для которых проводился расчет PDOS, отмечены на вставках как Au, Cu, Ni.
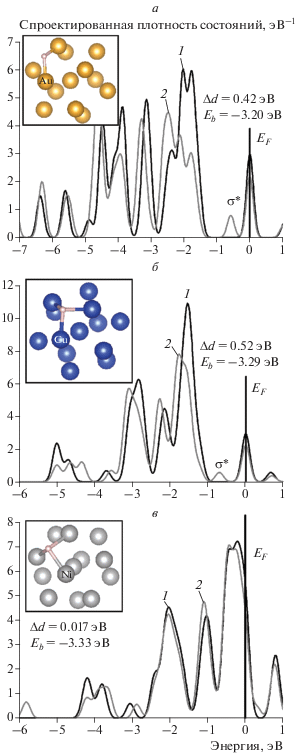
Кластеры AunNim, n + m = 13
Расчет изменений спроектированных плотностей состояний атомов кластера биметалла при адсорбции атома Н проводился для каждого из ряда кластеров Au12Ni1, Ni12Au1, jan-Au7Ni6, jan-Ni7Au6, Au7Ni6 для ближайшего к адсорбату атома и для соседнего атома (табл. 3). На рис. 3 представлены структуры адсорбционных комплексов на основе кластеров биметаллов Au12Ni1 и Ni12Au1 соответственно, а также отражено изменение электронного строения этих кластеров при адсорбции водорода на различных сайтах. Для кластера Au12Ni1 были выбраны два начальных положения атома Н (сайта, (s)): над атомом Ni (s1) и над ближайшим атомом Au (s2). По аналогии были заданы сайты атома Н для кластера Ni12Au1: над атомом Au (s1) и над ближайшим атомом Ni (s2). На рис. 3 показаны два примера рассчитанных спроектированных плотностей состояний для сайта s1 кластера Au12Ni1 (табл. 3, строка 5) и для сайта s1 кластера Ni12Au1 (табл. 3, строка 9). Как видно, трансформация энергетических спектров атомов Au сохраняет свою тенденцию.
Рис. 3.
Изменения PDOS кластеров AunNim (n + m = 13) при адсорбции атома H: а – на атоме Au кластера Au12Ni1, б – на атоме Ni кластера Au12Ni1, в – на атоме Au кластера Ni12Au1, г – на атоме Ni кластера Ni12Au1; 1 – PDOS1, 2 – PDOS2. На вставках к рисункам б и г приведены атомные структуры кластеров. Атомы, для которых проводился расчет PDOS, отмечены на вставках как Au и Ni.
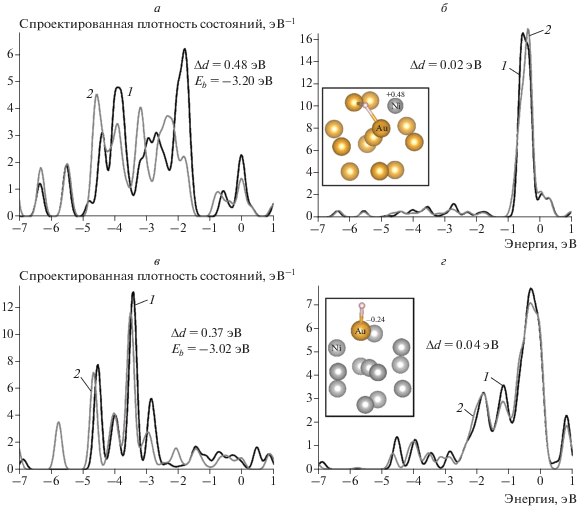
Два сайта атома Н для кластера Au7Ni6 представляют собой атом Ni, окруженный атомами Au (s1), и атом Au, окруженный атомами Ni (s2). Для янус-кластеров исследование изменений электронной структуры было более подробным и включало в себя сайты вблизи интерфейса (si(Au) и si(Ni)) и на максимальном удалении от него каждой из подсистем (sv(Au) и sv(Ni)) соответственно. Результаты, а именно энергии и длины связей, конечные сайты, центры d-зон и их изменения, представлены в табл. 3.
Суммируя данные из расчета электронной структуры и табл. 1 можно сделать следующие выводы. Электроотрицательность атома Ni, χNi = = 1.91, меньше электроотрицательности атома Au, χAu = 2.54 [17], поэтому в биметаллических кластерах атом Ni приобретает положительный заряд, а атом Au – отрицательный, что подтверждается расчетами заселенности по Малликену. из-за избытка положительного заряда адсорбция H в окрестности примесного атома Ni энергетически нестабильна. При этом примесный атом Au имеет отрицательный заряд, что способствует адсорбции атома H в его окрестностях. В кластере Au7Ni6 и янус-кластерах благодаря равномерному распределению атомов разного типа перетекание электронной плотности меньше, поэтому адсорбция атома H стабильна и в окрестности атомов Ni. Стоит отметить, что при адсорбции H электронная структура атомов биметаллического кластера качественно изменяется так же, как и в моноатомных кластерах Au13 и Ni13: происходит сдвиг центра d-зоны атомов Au и не происходит заметных изменений плотности состояний атомов Ni.
При взаимодействии атомов Ni и Au в биметаллических кластерах, атомная структура испытывает трансформацию вследствие того, что равновесное расстояние в кластере Ni13 меньше равновесного расстояния в кластере Au13. Это приводит к усилению перекрытия орбиталей атомов Au при уменьшении длин связей атомов кластера биметалла, что проявляется как уширение PDOS атомов Au. Перекрытие орбиталей атомов Ni, напротив, уменьшается при увеличении длин связей атомов кластера биметалла, что проявляется как сужение PDOS атомов Ni. Следствием такого изменения PDOS является тенденция к сдвигу вниз (от уровня Ферми) центра d-зоны атомов Au и сдвигу вверх (к уровню Ферми) центра d-зоны атомов Ni, что в соответствии с резонансной моделью хемосорбции проявляется как уменьшение энергии связи с H в окрестности атомов Au по сравнению с моноатомным кластером Au13 и увеличение величины энергии связи атома H в окрестностях атомов Ni по сравнению с моноатомным кластером Ni13. Отклонения от общей тенденции наблюдаются в окрестностях локальных неоднородностей атомной структуры и могут быть обусловлены нарушением общей симметрии кластера
Таким образом, полученный результат коррелирует с данными эксперимента по адсорбции H на биметаллическом наноструктурированном покрытии Au–Ni и подтверждает предположение о возможности накопления адсорбированного H на Ni-наночастицах без существенного изменения их электронного строения [10, 18]. Накопленный H может принимать участие в дальнейших химических превращениях. Уменьшение энергии связи с H в окрестности атомов Au может способствовать увеличению подвижности промежуточных продуктов реакции и уменьшению отравления побочными веществами.
Кластеры AunCum, n + m = 13
Расчет изменений спроектированных плотностей состояний атомов кластера биметалла при адсорбции атома Н проводился для каждого из ряда кластеров Au12Cu1, Cu12Au1, jan-Au7Cu6, jan-Cu7Au6, Au7Cu6 для ближайшего к адсорбату атома и для соседнего атома (табл. 3). На рис. 4 представлены структуры адсорбционных комплексов на основе кластеров биметаллов Au12Cu1 и Cu12Au1 соответственно, а также отражено изменение электронного строения этих кластеров при адсорбции водорода на различных сайтах.
Рис. 4.
Изменения PDOS кластеров AunCum (n + m = 13) при адсорбции атома H: а – на атоме Au кластера Au12Cu1, б – на атоме Cu кластера Au12Cu1, в – на атоме Au кластера Cu12Au1, г – на атоме Cu кластера Cu12Au1; 1 – PDOS1, 2 – PDOS2. Во вставках к рисункам б и г приведены атомные структуры кластеров. Атомы, для которых проводился расчет PDOS, отмечены на вставках как Au и Cu.
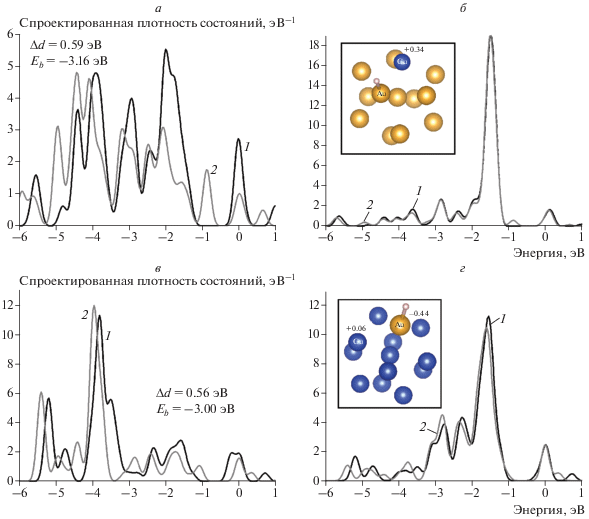
Для кластера Au12Cu1 были выбраны два начальных сайта атома Н: над атомом Cu (s1) и над ближайшим атомом Au (s2). Поскольку конечные значения энергии и длины связи, а также изменения энергетических спектров для обоих сайтов имели очень близкие величины, их объединили в один. По аналогии были заданы сайты атома Н для кластера Cu12Au1: над атомом Au (s1) и над ближайшим атомом Cu (s2). На рис. 4 показаны два примера рассчитанных спроектированных плотностей состояний для сайта s1 кластера Au12Cu1 (табл. 3) и для сайта s1 кластера Cu12Au1 (табл. 3). Как видно, трансформация энергетических спектров атомов Au сохраняет свою тенденцию.
Два сайта атома Н для кластера Au7Cu6 представляют собой атом Au, окруженный атомами Cu (s1), и атом Au, окруженный атомами Cu (s2). Для янус-кластеров исследование изменений электронной структуры было более подробным и включало в себя сайты вблизи интерфейса (si(Au) и si(Cu)) и на максимальном удалении от него каждой из подсистем (sv(Au) и sv(Cu)) соответственно. Результаты, а именно энергии и длины связей, конечные сайты, центры d-зон и их изменения, представлены в табл. 3.
Суммируя данные из расчета электронной структуры и табл. 3, можно сделать следующие выводы. Электроотрицательность атома Cu χCu = 1.90 [17], что, так же, как и электроотрицательность атома Ni, меньше электроотрицательности атома Au, поэтому в биметаллических кластерах атом Cu приобретает положительный заряд, а атом Au – отрицательный, что подтверждается расчетами заселенности по Малликену. В целом, перенос заряда между подсистемами Au и Cu меньше, чем между подсистемами Au и Ni. Адсорбция H энергетически нестабильна в окрестности примесного атома Cu и стабильна в окрестности примесного атома Au, по аналогии с никелем. В кластере Au7Cu6 и янус-кластерах, благодаря равномерному распределению атомов разного типа, перетекание электронной плотности меньше, поэтому адсорбция атома H стабильна и в окрестности атомов Cu. Стоит отметить, что при адсорбции H электронная структура атомов биметаллического кластера качественно изменяется так же, как и в моноатомных кластерах Au13 и Cu13: происходит сдвиг центра d-зоны атомов Au и Cu.
Трансформация атомной структуры в кластерах Cu–Au несколько меньше, чем в кластерах Au–Ni, однако имеет схожие тенденции, поскольку равновесное расстояние в кластере Cu13 меньше равновесного расстояния в кластере Au13. Это также приводит к усилению перекрытия орбиталей атомов Au и уменьшению перекрытия орбиталей атомов Cu, следовательно PDOS атомов Au расширяются, а PDOS атомов Cu сужаются. Следствием такого изменения PDOS является тенденция к сдвигу вниз (от уровня Ферми) центра d-зоны атомов Au и сдвигу вверх (к уровню Ферми) центра d-зоны атомов Cu, что в соответствии с резонансной моделью хемосорбции проявляется как уменьшение энергии связи с H в окрестности атомов Au по сравнению с моноатомным кластером Au13 и увеличение величины энергии связи атома H в окрестностях атомов Cu по сравнению с моноатомным кластером Cu13. Однако для атомов Cu проявляется отклонение от резонансной модели хемосорбции; центры d-зон атомов Cu при этом сдвигаются вниз, что в данном случае может быть обусловлено вкладом поверхностных состояний вблизи уровня Ферми и переносом заряда в системе, причем оба фактора могут быть взаимосвязаны.
Таким образом, полученный результат коррелирует с данными эксперимента по адсорбции атома H на наночастицах биметалла Au–Cu, в котором наблюдалось заметное изменение электронной структуры наночастиц меди [19, 20]. Уменьшение энергии связи с H в окрестности атомов Au может способствовать увеличению подвижности промежуточных продуктов реакции и уменьшению отравления побочными веществами.
ЗАКЛЮЧЕНИЕ
В результате квантовохимического моделирования предсказаны эффекты взаимодействия Au–Ni и Au–Cu кластеров с атомарным водородом. Установлено, что вследствие изменения расстояний между атомами и перераспределения электронной плотности в биметаллических кластерах энергия связи атомов Н уменьшается в окрестности атомов Au и увеличивается в окрестностях атомов Ni/Cu, что может привести к накоплению водорода на подсистеме Ni/Cu и улучшить реакционную способность подсистемы Au, поскольку промежуточным реагентам или побочным продуктам будет проще вступить в реакцию или десорбироваться с поверхности. Результаты численных экспериментов коррелируют с экспериментальными данными по адсорбции водорода на биметаллических наноструктурированных покрытиях золото–никель и золото–медь, исследованных с помощью СТМ.
Все расчеты выполнены с использованием вычислительных ресурсов Межведомственного суперкомпьютерного центра РАН (МСЦ РАН).
Работа выполнена в рамках государственного задания № 0082-2014-0011 “Нанохимия” (АААА-А17-117111600093-8), а также частичного финансирования Российским фондом фундаментальных исследований (гранты № 17-03-00275, № 18-03-00060).
Список литературы
Tsai C.-H., Xu M., Kunal P. et al. // Catal. Today. 2018. V. 306. P. 81.
Wang Q., Wang W., Yan B. et al. // Chem. Eng. J. 2017. V. 326. P. 182.
Gao W., Li F., Huo H. et al. // J. Mol. Catal. 2018. V. 448. P. 63.
Groß A. // Top. Catal. 2006. V. 37. № 1. P. 29.
Shi J. // Chem. Rev. 2013. V. 113. № 3. P. 2139.
Yuan D. W., Wang Y., Zeng Zh. // J. Chem. Phys. 2005. V. 122. P. 114310.
Nigam S., Sahoo S.K., Sarkar P. et al. // Chem. Phys. Lett. 2013. V. 584. P. 108.
Ma L., Laasonen K., Akola J. // J. Phys. Chem. C. 2017. V. 121. № 20. P. 10876.
Дохликова Н.В., Колченко Н.Н., Гришин М.В. и др. // Рос. Нанотехнол. 2016. Т. 11. № 1–2. С. 6.
Гришин М.В., Гатин А.К., Дохликова Н.В. и др. // Хим. физика. 2019. Т. 38. № 1.
Giannozzi P., Baroni S., Bonini N. et al. // J. Phys.: Condens. Matter. 2009. V. 21. P. 395502.
Ozaki T., Kino H. // Phys. Rev. B. 2004. V. 69. № 19. P. 195113.
Cors A.D. // Comput. Mater. Sci. 2014. V. 95. P. 337.
Гилл Ф., Мюррей У., Райт М. Практическая оптимизация. Пер. с англ. М.: Мир, 1985.
Hammer B. // Top. Catal. 2006. V. 37. № 1. P 3.
Hammer B., Norskov J.K. // Nature. 1995. V. 376. P. 238.
Pauling L. The nature of the chemical bond. 3 ed. N.Y.: Ithaca, 1960.
Кирсанкин А.А., Гришин М.В., Сарвадий С.Ю. и др. // Хим. физика. 2017. Т. 36. № 6. С. 79.
Gatin A.A., Grishin M.V., Dokhlikova N.V. et al. // Nanotechnol. Rus. 2018. V. 13. № 9–10. P. 453.
Гатин А.К., Гришин М.В., Сарвадий С.Ю. и др. // Хим. физика. 2018. Т. 37. № 3. С. 18.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика