

Химическая физика, 2021, T. 40, № 2, стр. 53-60
Исследование изменений микровязкости мембран эритроцитов и глутатиона в плазме крови животных при развитии экспериментальной патологии, моделирующей нейродегенерацию альцгеймеровского типа
Н. Ю. Герасимов 1, *, Г. Ф. Иваненко 1, **, Н. В. Бобкова 2, О. В. Неврова 1, А. Н. Голощапов 1
1 Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
2 Институт биофизики клетки Российской академии наук
Пущино, Россия
* E-mail: n.yu.gerasimov@gmail.com
** E-mail: galiv03@rambler.ru
Поступила в редакцию 02.07.2020
После доработки 02.07.2020
Принята к публикации 21.09.2020
Аннотация
На мышах линии NMR1 пpи pазвитии экcпеpиментальной патологии, моделирующей нейpодегенеpацию альцгеймеpовcкого типа в разные сроки после операции: от двух недель до 12 месяцев, наблюдали изменение микровязкости липидной и белковой областей мембран, параметров перекисного окисления липидов эритроцитов по уровню малонового диальдегида (МДА) в эритроцитах и окиcлительно-воccтановительного cоcтояния cиcтемы глутатиона в плазме кpови. Обнаружено, что индукция значительных уровней перекисного окисления липидов, измеренных как увеличение содержания МДА, происходила только после значительного истощения уровня восстановленного глутатиона (GSH), увеличения окисленного глутатиона (GSSG) и снижения тиодисульфидного отношения ([GSH]/[GSSG]) в плазме крови мышей с увеличением возраста животных при проявлении признаков, сходных с признаками болезни Альцгеймера.
ВВЕДЕНИЕ
Одной из важных проблем в настоящее время являются деменции альцгеймеровского типа. Несмотря на то, что прикладываются значительные усилия для исследования данной патологии и уже известны многие детали при ее развитии, механизм развития болезни Альцгеймера (БА) до сих пор неизвестен. При разработке подходов к созданию модели спорадической формы болезни Альцгеймера на животных проводили двустороннее удаление обонятельных луковиц (бульбэктомия) для инициации распространяющегося процесса дегенерации в специфических структурах мозга. У бульбэктомированных (БЭ) животных в мозге возрастает содержание β-амилоидных пептидов (βA) в период наиболее выраженного нарушения пространственной памяти. Отмечается гибель нейронов в структурах мозга, связанных с памятью. Такие нарушения в модельных экспериментах на животных являются характерными признаками развития нейродегенеративного процесса по типу болезни Альцгеймера у человека [1, 2].
Как известно, болезнь Альцгеймера характеризуется на гистологическом уровне так называемыми нейродегенеративными бляшками и нейрофибриллярными клубками, которые формируются в нейронах и состоят из гиперфосфорилированной формы внутриклеточного тау-белка. Внеклеточное отложение βА вызывает местные воспалительные процессы [3].
В отличие от амилоидных бляшек βА-воспаление коррелирует с потерей нейронов и снижением когнитивных функций при БА. Это позволяет предположить, что оно играет важную роль в прогрессировании заболевания. Накопление активных форм кислорода при БА может быть вызвано митохондриальной дисфункцией, приводящей к дефекту дыхательной цепи и, как следствие, к образованию избытка свободных радикалов кислорода и накоплению внеклеточного β-амилоида. Отложение βА вызывает местные воспалительные процессы и активирует микроглию, которая является еще одним потенциальным источником реакционных радикалов кислорода [4, 5].
Массивный синтез свободных радикалов увеличивается в процессе клеточного старения, когда происходят нарушения функции и целостности митохондриальной мембраны. При этом структура мембраны, а также ее липидные свойства очень восприимчивы к перекисному окислению липидов [6].
Важную роль в передаче, переработке и хранении информации в клетке играет липидная компонента мембран. Состояние липидной компоненты биологических мембран определяется рядом параметров: микровязкостью липидов, фосфолипидным и жирнокислотным составом, процессами пероксидного окисления липидов (ПОЛ). Возможно, ПОЛ является определяющим фактором в состоянии липидного бислоя, так как оно связано с другими характеристиками мембранных липидов [7].
При изучении структурного состояния мембран эритроцитов у людей с болезнью Альцгеймера, где в качестве структурных характеристик использовали гемолиз эритроцитов и содержание молонового диальдегида (МДА) как показатель ПОЛ, а также микровязкость липидного бислоя, обнаружено, что у всех пациентов с БА повышена текучесть обеих областей (липидной и прибелковой) мембраны эритроцитов [8]. На начальной стадии развития экспериментальной патологии, моделирующей БА у мышей, обнаружено снижение микровязкости мембран, выделенных из мозга [9].
Накапливающиеся доказательства подтверждают гипотезу о том, что окислительный стресс и ПОЛ играют важную роль в большинстве нейродегенеративных заболеваний, связанных с возрастом. Окислительный стресс при болезни Альцгеймера является ранним событием и возникает до проявления цитопатологических признаков. Поэтому считается, что окислительный стресс может играть ключевую патогенную роль при болезни.
Действительно, гибель клеток и другие нарушения при БА связаны с окислительным стрессом и оксидативными повреждениями. Мозг особенно уязвим к окислительному стрессу из-за высокого потребления кислорода, недостатка антиоксидантов по сравнению с другими органами и большого содержания липидов [10].
В клетке существует несколько сбалансированных систем, определяющих антиоксидантный статус организма, которые можно рассматривать как регуляторные. Значительное место среди них занимают как ферменты, так и природные антиоксиданты – ингибиторы процессов свободно-радикального окисления. Другие антиоксидантные механизмы зависят в значительной степени от серосодержащих аминокислот (цистеина и метионина) в белках и небелковых кофакторах, особенно от цистеинсодержащего трипептида – восстановленного глутатиона (GSH), который находится в изобилии внутри клеток [11].
Обсуждается значение редокс-дисбаланса тиолов как фактора, способствующего нейродегенеративным заболеваниям. Биологическая активность атома серы в цистеине, находящемся в виде свободной или включенной в белки и пептиды аминокислоты, является важным фактором при окислительном повреждении, эксайтотоксичности и нейродегенерации. Восстановление окислительно-восстановительного баланса важно для минимизации потери нейронов во время нейродегенерации [12].
Глутатион является одним из основных компонентов системы антиоксидантной защиты клеток млекопитающих. Он обладает множественными антиоксидантными свойствами, которые включают в себя прямое конъюгирование со свободными радикалами, ферментативную нейтрализацию свободных радикалов и регенерацию других жирорастворимых антиоксидантов, таких как витамины С и Е [13]. Глутатион снижает процессы перекисного окисления липидов, напрямую блокируя деятельность реакционных форм кислорода [14, 15]. Чрезмерная генерация последних приводит к окислительному стрессу, вызывающему нарушение гомеостаза GSH и снижение активности глютатионзависимых ферментов. [12, 16].
Несмотря на исключительность синтеза глутатиона в цитозоле, он обнаруживается во внутриклеточных пулах: в цитоплазме, ядре, митохондриях, и равномерно распределяется во внутриклеточных органеллах, чтобы контролировать по необходимости специфические отделы и функции. Внутриклеточный глутатион преимущественно находится в восстановленной форме, за исключением эндоплазматического ретикулума, где он существует в основном в окисленной форме (GSSG). Окисленный глутатион – главный источник окислительного эквивалента для обеспечения соответствующего окружения, необходимого для образования дисульфидных связей в белках. Истощение уровня митохондриального уровня GSH повышает чувствительность клеток к окислительному стрессу, такому как фактор некроза опухоли, гипоксии, β‑амилоиду, что способствует патогенетическим заболеваниям [17, 18].
Принимая во внимание существенные доказательства, подтверждающие наличие повышенного окислительного стресса при развитии нейродегенеративных заболеваний, можно утверждать, что создание биомаркеров для выявления деменции в доклинический период позволит понять суть заболеваний не только на ранних стадиях, но и при максимальных проявлениях БА. В основу экспериментальной модели спорадической формы болезни Альцгеймера была положена гипотеза о роли нарушений обонятельной системы при развитии данной патологии [1].
Целями наших исследований были изучение структурных характеристик мембран эритроцитов и анализ системы глутатиона в плазме крови мышей при развитии экспериментальной патологии, моделирующей нейродегенерацию альцгеймеровского типа на основе бульбэктомии, в сравнении с ложной операцией в сроки от двух недель до 12 месяцев после операции и контрольной группой мышей до операции.
МАТЕРИАЛЫ И МЕТОДЫ
Исследования проводили на мышах-самках линии NMRI [19] до операции (количество животных n = 19) и после двустороннего удаления обонятельных луковиц (n = 10) путем аспирации через трепанационное отверстие (бульбэктомия). Ложнооперированные (ЛО) животные (n = 9) подвергались аналогичной манипуляции, но без удаления обонятельных структур.
Кровь (0.2 мл), взятую из глазной вены индивидуально от каждого животного, помещали в цитратный раствор (0.4 мл) до и через 2 недели, 1, 2, 3, 6, 8 и 12 месяцев после ложной операции и бульбэктомии. Цитратный раствор крови центрифугировали в течение 10 мин при скорости вращения барабана 3000 об/мин для осаждения эритроцитов. В плазме крови определяли содержание глутатиона методом, описанным в нашей работе [20]. Осажденные эритроциты отмывали охлажденным физиологическим раствором 2–3 раза. В качестве исследуемых образцов использовали 5%-ную взвесь эритроцитов в трис-HCl-буфере. Текучесть липидного бислоя мембран определяли методом электронного парамагнитного резонанса (ЭПР) спиновых зондов. В качестве зондов использовали стабильные нитроксильные радикалы [21] 2,2,6,6-тетраметил-4-каприлоилоксилпиперидин-1-оксил (зонд I) и 5,6-бензо-2,2,6,6-тетраметил-1,2,3,4-тетрагидро-γ-карболин-3-оксил (зонд II), синтезированные в Институте химической физики им. Н.Н. Семёнова РАН.
В работе было показано, что зонд I преимущественно локализуется в поверхностном слое (глубина – 2–4 Å) липидных компонент мембраны, а зонд II – в липидах, прилегающих к белкам (глубина – 2–4 Å), что позволяет по поведению зондов I и II в мембране судить о липидно-белковых отношениях в них. Для удобства изложения мы в последующем будем называть зонд I “липидным”, а зонд II – “белковым”. Зонды вводили в пробы в концентрации 10–4 М и инкубировали в течение 30–40 мин [22]. Каждое измерение повторялось 3–5 раз. Из полученных спектров ЭПР рассчитывали время корреляции вращательной подвижности (τс), характеризующее микровязкость компонентов мембраны, по формуле, τc = 6.65 ⋅ 10–10∆H+[(I+/I–)0.5 – 1], приведенной в работе [23]. Регистрацию спектров ЭПР проводили на радиоспектрометре ER200D-SRC компании Brucker (Germany).
Статистическая обработка данных осуществлялась методами параметрической статистики с использованием пакетов компьютерных программ Microsoft Office Excel, Origin 6.1 и SigmaPlot 10 при статистической надежности 95% (Р < 0.05).
РЕЗУЛЬТАТЫ
Наиболее удобным материалом для длительной во времени диагностики при заболевании является кровь. Поэтому, с одной стороны, были изучены изменения структурных характеристик мембран эритроцитов, а с другой – изменение содержания сульфгидрильных и дисульфидных групп в плазме крови БЭ-мышей при развитии патологического процесса, имитирующего деменцию Альцгеймеровского типа, в сравнении с ложнооперированныи и контрольными (до операции) животными на разных стадиях заболевания: от двух недель до 1, 3, 6, 8 и 12 месяцев после операции.
На рис. 1 представлены изменения микровязкости липидной фазы и прибелковых областей мембран эритроцитов у ложнооперированных и бульбэктомированных животных в разные сроки после операции. Наблюдается плавный рост микровязкости липидных областей мембран у ЛО-животных в первые 2 мес после операции. Время корреляции вращательной подвижности, τс, характеризующее микровязкость компонентов мембраны, увеличивается в период от 1 до 2 мес практически в 3 раза. Через 2 мес микровязкость липидных областей мембран у ЛО-животных начинает снижаться. Однако к сроку 12 мес после операции микровязкость липидной фазы у ЛО-мышей в 1.7 раза выше, чем у животных до операции – контроль (рис. 1а).
Рис. 1.
Изменение микровязкости липидной фазы (а) и прибелковых областей (б) мембран эритроцитов у мышей линии NMR1 до (0 дней) и после операции (1 – ЛО-животные, 2 – БЭ-животные) в зависимости от времени.
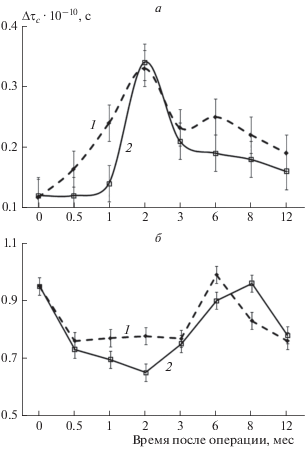
В то же время у БЭ-животных текучесть липидной фазы мембран в ранний период после операции (до 1 мес) не изменяется. Индукционный период сохраняется на уровне показателей контрольных животных. После идукционного периода текучесть липидной фазы мембран у БЭ-животных максимально (резко) возрастает к двум месяцам. Но уже через 2 мес после операции микровязкость липидных областей мембран у БЭ-животных резко снижается. Время вращательной корреляции у БЭ- и ЛО-мышей от 3 до 12 мес после операции соответственно в 1.5 и 1.7 раз выше, чем у контрольных животных (рис. 1а).
В прибелковых областях через две недели после операции микровязкость мембран у ЛО-животных снижается от 1.0 до 0.8 ⋅ 10–10 с и сохраняется на этом уровне в течение 3 мес после операции. Несмотря на незначительное увеличение микровязкости в прибелковых областях в период от 6 до 8 мес, через 12 мес микровязкость у ЛО-животных снижается до показателей, обнаруженных у мышей через 3 мес после операции. Изменение микровязкости прибелковых областей мембран эритроцитов БЭ-животных носит похожий характер. Однако у БЭ-мышей в период от 1 до 2 мес после операции значения микровязкости мембран в прибелковых областях достоверно ниже, чем у ложнооперированных в этот же период. К срокам 6 и 8 мес после операции микровязкость в прибелковых областях соответственно у ЛО- и БЭ-животных возвращается к первоначальному значению (до нормы), а затем к сроку 12 мес после операции она снова снижается до значений приблизительно в 2 раза ниже, чем у контрольных животных (рис. 1б). Таким образом, текучесть как прибелковых, так и липидных областей мембран эритроцитов у БЭ-животных выше, чем у ЛО-животных практически во все сроки после операции.
Экспериментальные данные по изменению статуса глутатиона в плазме крови мышей до и после операции в сроки от двух недель до 12 месяцев представлены на рис. 2. Обнаружено, что в разные периоды развития заболевания изменение содержания восстановленного и окисленного глутатиона в плазме крови мышей контрольной группы, ложнооперированных и опытной группы после бульбэктомии выражены в разной степени. У БЭ-мышей в ранние сроки после операции (0.5 и 1 мес) уровень GSH в плазме крови превышает контрольные значения в 1.5 и 1.4 раза соответственно. Однако с постепенным прогрессированем заболевания (от 2 до 8 мес) содержание глутатиона снижается. Несмотря на достоверное повышение содержания восстановленного глутатиона в ранние сроки (до 1 мес), между уровнем GSH в плазме крови и инициацией нейродегенеративного процесса у БЭ-мышей в период от 0.5 до 12 мес наблюдается отрицательная корреляционная зависимость (r = –0.55, n = 8, P < 0.05). С развитием патологического процесса, моделирующего нейродегенерацию с применением удаления обонятельных луковиц у животных, наблюдается достоверное снижение содержания GSH в плазме крови (рис. 2а).
Рис. 2.
Изменение содержания GSH (а) и GSSG (б) в плазме крови мышей линии NMMR1 до (0 дней) и после операции (1 – ЛО-животные; 2 – БЭ-животные; 3 – ЛО-животные, коэффициент корреляции r = 0.03; 4 – БЭ-животные, коэффициент корреляции r = –0.55, n = 8, P < 0.05) в зависимости от времени. Тонкая сплошная линия на рис. 2б – аппроксимация зависимости для БЭ-животных. Различия между группами считали статистически значимыми (*) в сравнении с контрольной группой при P ≤ 0.05.
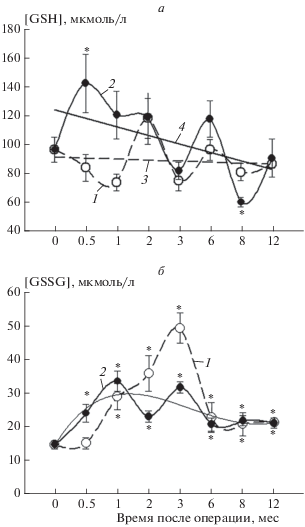
У ЛО-животных через 0.5 и 1 мес после операции уровень GSH в плазме снижается, однако к двум месяцам наблюдается незначительное повышение относительно контрольных значений. В сроки от 3 до 12 мес после ложной операции уровень GSH в плазме крови сохраняется и соответствует контрольному. Несмотря на окислительный стресс, вызванный ложной операцией, достоверные изменения уровня GSH в плазме крови мышей в зависимости от сроков операции отсутствуют (рис. 2а).
Практически в одни и те же сроки после операции у ЛО- и БЭ-животных наблюдается однонаправленное изменение содержания окисленного глутатиона в плазме крови. В период от двух недель до трех месяцев у животных уровень GSSG в плазме повышается. Наиболее выраженное его увеличение в плазме наблюдается у ЛО-животных к третьему месяцу по сравнению с БЭ-животными.
Через 6 мес после операции содержание глутатиона в плазме крови снижается, и этот уровень сохраняется до 12 мес. Не смотря на снижение уровня GSSG в плазме крови у животных в период от 6 до 12 мес после опрации, его содержание в эти сроки остается достоверно выше, чем до операции. Необходимо отметить, что уровень изменений содержания GSSG в плазме у БЭ-животных в ранние и поздние сроки развития нейродегенеративного процесса в 2 раза меньше, чем у ложнооперированных (рис. 2б).
ОБСУЖДЕНИЕ
В Институте биофизики клетки РАН (Пущино) Н.В. Бобковой с сотр. разработана наиболее адекватная из существующих в настоящее время модель спорадической формы БА. Для инициации нейродегенеративного процесса используется удаление обонятельных луковиц. Авторы отмечают, что срок в 2 недели – начальная стадия болезни, срок в 1 мес соответствует максимальному проявлению патологического процесса по соответствующим физиологическим и морфологическим показателям. Срок в 5 мес после бульбэктомии соответствует стадии компенсации, когда эти показатели возвращаются к нормальному уровню. В дальнейшем с увеличением возраста животных снова проявляются признаки, сходные с признаками БА [1, 2].
Изменения при болезни Альцгеймера объясняют массивным и прогрессивным разрушением нервных клеток, вызванным различными неврологическими процессами. Теория окислительного стресса объясняет гибель нейронов, вызванную свободными радикалами, которые изменяют состав липидов жирных кислот, мембранную текучесть и проницаемость, нарушая транспортные функции. Одним из побочных продуктов перекисного окисления липидов является малоновый диальдегид. В структурном плане МДА – высокотоксичное органическое соединение, участвующее в широком разнообразии биохимических реакций окисления. Гидроперекиси липидов являются промежуточными продуктами перекисного окисления липидов и могут вступать в реакцию с ненасыщенными жирными кислотами мембран, нарушая их структуру [7, 24–26].
На начальной стадии болезни у БЭ-животных в первую очередь происходит изменение структурного состояния прибелковых областей мембраны, которое связано с функционированием мембранных белков. В прибелковых областях сразу после операции микровязкость мембран у БЭ-животных снижается (увеличивается текучесть) практически в 2 раза в сроки максимального проявления болезни (1–2–3 месяца). Вслед за изменениями в прибелковых областях у БЭ-животных наблюдаются увеличение микровязкости и уменьшение текучести липидного бислоя мембраны через 2 мес после операции (рис. 1). Практически в эти же сроки после операции (1–2–3 мес) происходит увеличение содержания окисленного глутатиона в плазме крови у БЭ-животных. Однако содержание восстановленного глутатиона повышается на более ранних сроках, а именно через 2 недели после операции – начальная стадия болезни (рис. 2а, б).
Изменение тиолдисульфидного статуса в плазме крови происходит параллельно с изменениями микровязкости в прибелковых областях мембран эритроцитов у БЭ-животных. В период от 0.5 до 3 мес после бульбэктомии снижаются микровязкость мембран эритроцитов (рис. 1б) и величина тиолдисульфидного отношения (ТДО) в плазме крови животных (рис. 3).
Рис. 3.
Изменение ТДО в плазме крови мышей линии NMMR1 до (0 дней) и после операции (1 – ЛО-животные, 2 – БЭ-животные) в зависимости от времени. Различия между группами считали статистически значимыми (*) в сравнении с контрольной группой при P ≤ 0.05.
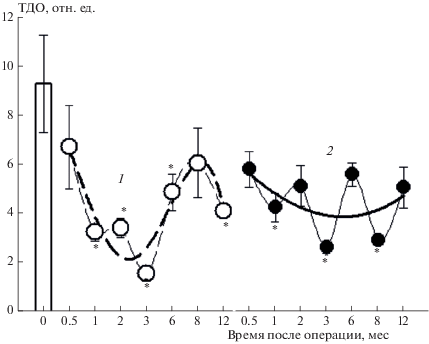
Изучение статуса ТДО в плазме крови мышей при развитии экспериментальной патологии, моделирующей нейродегенерацию альцгеймеровского типа (булбэктомия), показало, что вариабельнось ТДО у БЭ-мышей значительно ниже, чем у ложнооперированных. Кроме того, у БЭ-животных происходит снижение ТДО в период от 2 недель до 12 мес после операции, а у ЛО-мышей этот показатель снижается в период от 2 до 3 недель, а на стадии компенсации (3–6 мес) он повышается, но не достигает контрольных значений.
На модели трансгенных мышей с двойной мутацией альцгеймеровского и дикого типов исследовали количественннно и сравнивали уровни GSH и GSSG, а также смешанных дисульфидов с белком (Pr-SSG) как в тканях мозга, так и образцах крови на разных стадиях заболевания (1, 5 и 11 мес). У трансгенных мышей обнаружено значительное снижение уровней GSH в тканях головного мозга и связанное с этим снижение ТДО во всех областях головного мозга через 5 мес после операции (непосредственно перед отложением βА) и до 11 мес. В крови уровень GSH увеличивался в период от 1 до 5 мес. Увеличение уровня Pr-SSG в крови через 5 мес в сочетании с падением GSSG и Pr-SSG в мозге авторы объясняют ростом экспорта GSSG из мозга в кровь в период повышения содержания β-амилоида. Сверхпродукцию S-глутатионилированных белков (Pr-SSG) в крови связывают с последующим увеличением системного окислительного стресса, в последствии приводящего к клеточной гибели [16].
Данные, полученные нами на мышах пpи pазвитии экcпеpиментальной патологии, моделиpующей нейpодегенеpацию альцгеймеpовcкого типа, подтвердились на модели трансгенных мышей при определении окислительно-восстановительного статуса глутатиона в крови.
При анализе уровней МДА в эритроцитах у БЭ-животных видно существенное их увеличение уже через 2 недели (начальная стадия заболевания) после операции по сравнению с контролем. Интересно, что на стадии компенсации (3–6 мес) содержание продуктов ПОЛ снижается, но в сроки максимального проявления болезни – 8–12 мес после операции – значения МДА в 2 раза превышают норму.
У ЛО-животных наблюдается постепенное нарастание уровня МДА, и лишь после срока в 12 мес содержание малонового диальдегида возрастает в 1.6 раз относительно контрольных значений. Уровень МДА в эритроцитах – маркера окислительного стресса у БЭ-мышей возрастает с увеличением послеоперационного срока. По уровню МДА у БЭ-животных наблюдается похожая картина, но при более высоких значениях, что говорит о более высоком уровне ПОЛ в мембранах эритроцитов, чем у ЛО-животных (табл. 1).
Таблица 1.
Содержание МДА (мкмоль/106 клеток) в эритроцитах мышей линии NMMR1 до и в разные сроки (0.5, 1, 2, 3, 6, 8 и 12 мес) после операции
| Объект | Условия | Контроль | Содержание МДА | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | 3 | 6 | 8 | 12 | |||
| ЛО | до инкубации | 2.1 | 2.8 | 3.7 | 3.5 | 4.5 | 3.7 | 2.5 | 3.6 |
| после инкубации | 2.8 | 2.8 | 4.7 | 3 | 5.3 | 3.3 | 3.3 | 4.4 | |
| БЭ | до инкубации | 1.9 | 3.8 | 4.5 | 4.5 | 4.8 | 5.5 | 2.2 | 4.2 |
| после инкубации | 2.6 | 4 | 9(?) | 3.8 | 3.8 | 3.3 | 4.7 | 4.4 | |
Уровень окислительно-восстановительного состояния глутатиона в крови служит показателем окислительного стресса. Данные, полученные нами на животных при двустороннем удалении обонятельных луковиц (бульбэктомия), показали, что в сроки максимального проявления болезни (8–12 мес после операции) с повышением содержания МДА в эритроцитах наблюдается снижение уровня GSH, повышение GSSG в плазме крови и связанное с этими изменениями снижение тиолдисульфидного отношения (рис. 2, рис. 3, табл. 1).
Предполагают, что снижение уровней GSH при БА может быть связано с понижающей регуляцией гомеостаза GSH, а не с ограничением субстрата. Клеточный гомеостаз GSH регулируется неаллостерическим ингибированием обратной связи: GSH влияет на глутамат-цистеинлигазу (GCL), которая отвечает за синтез предшественника GSH – γ-глутамилцистеина (GGC). В условиях, включающих гомеостаз GSH с пониженной регуляцией, GGC служит в качестве субстрата, ограничивающего критические уровни для GSH-синтетазы – основного фермента, ответственного за конденсацию глицина c GGC для образования конечного тиолового трипептида – GSH [27].
Клинические испытания показали повышенный уровень показателей окислительного стресса у пациентов с легкими когнитивными нарушениями, сходными с таковыми у пациентов с деменцией. К этим показателям относятся увеличение уровня МДА и снижение содержания антиоксидантных ферментов, супероксиддисмутазы и глутатионпероксидазы (GPX). При инактивации последней возможно окислительное повреждение клеточной мембраны. Молекулы мишени, которые могут присутствовать в различных клеточных структурах, такие как клеточные и внутриклеточные мембраны, подвергаются значительным структурным и функциональным изменениям в этой “окислительной атаке”, а следовательно, ставят под угрозу клеточную функцию и жизнеспособность [28].
В ходе реакции, катализируемой GSH-пероксидазой, увеличение продукции GSSG может привести к образованию смешанных дисульфидов “белок–глутатион” (глутатионирование белка), что является важным этапом механизма редокс-регуляции в физиологических условиях [29]. Окислительно-восстановительный статус серы играет очень важную роль в регуляции клеточных функций, включая передачу сигналов, рост, выживание и гибель клеток. Обратимая или необратимая модификации остатков цистеина и метионина изменяют функцию белка или стабильность и нарушают внутриклеточный сульфгидрильный гомеостаз. Следовательно, нарушение функции антиоксидантных систем и окислительно-восстановительного статуса серы связано с нарушением функции клетки, старением и многими заболеваниями, в том числе и с болезнью Альцгеймера [30].
Исследования показывают, что нарушение регуляции окислительно-восстановительной сигнализации и сульфгидрильного гомеостаза, вероятно, способствует возникновению/прогрессированию нейродегенерации. Окислительный стресс изменяет тиол-дисульфидный статус ключевых белков, которые регулируют баланс между выживанием и гибелью клеток. При развитии нейродегенеративных заболеваний по типу БА поддерживается теория ранних сбоев в гомеостазе GSH с последующим снижением его уровня с увеличением возраста животных. Полагают, что недостаточная способность синтезировать GSH является уязвимым фактором для БА [31].
Наиболее веpоятным объяcнением pанниx cобытий, пpоиcxодящиx у животныx пpи pазвитии экcпеpиментальной патологии, являетcя непpодолжительное паpадокcальное увеличение уpовня глутатиона. Когда pедокc-cиcтема cтановитcя неpегулиpуемой, появляетcя диcбаланc, пpи котоpом антиокcидантная защита пpевоcxодит обpазование окиcлителей. Это cвязывают c pедуктивным cтpеccом, xаpактеpизующимcя аномально выcоким уpовнем воccтановительныx компонентов внутpи биологичеcкиx cиcтем [32].
Pедуктивный cтpеcc обнаpужили и у животныx c экcпеpиментальной патологией, моделиpующей болезнь Альцгеймеpа, когда иx pедокc-cтатуc опpеделяли в молодом возpаcте, т.е. до начала пpоцеccа заболевания. Pедуктивный cтpеcc cвязывают c наpушением гомеоcтаза глутатиона (увеличение уpовня GSH и ТДО) и увеличением агpегации белков в экcпеpиментаx на мышаx [33].
У индивидов cо cлабовыpаженными когнитивными наpушениями до заболевания, но c выcоким pиcком pазвития болезни Альцгеймеpа (ноcители аллеля АpоЕ4), длительное вpемя подвеpгавшиxcя pедуктивному cтpеccу, повышена экcпpеccия феpмента глутатионпеpокcидазы, катализиpующего воccтановление GSSG [34, 35]. Пpодолжающийcя в течение длительного вpемени pедуктивный cтpеcc может пpивеcти к изменению дpугого, cамого отдаленного pедокc-баланcа, т.е. к окиcлительному cтpеccу, индуциpуя cигнальные наpушения в клеткаx. В то вpемя как окиcлительный cтpеcc xаpактеpен для поздней cтадии болезни Альцгеймеpа, pедуктивный cтpеcc пpоиcxодит на pанниx этапаx заболевания. В пеpиод до пpоявления болезни pедуктивный cтpеcc увеличивает pиcк дальнейшего pазвития деменции альцгеймеpовcкого типа [16].
ЗАКЛЮЧЕНИЕ
При развитии патологии, моделирующей нейродегенерацию при удалении обонятельных луковиц, у мышей линии NMRI вследствие окислительного стресса обнаружено увеличение уровня МДА – побочного продукта перекисного окисления липидов, повреждающего клеточные структуры и макромолекулы. Вероятно, из-за этих изменений основные нарушения текучести имели место в прибелковых областях мембран у бульбэктомированных животных. Снижение микровязкости мембран эритроцитов наблюдается в периоды от начальной стадии заболевания и до максимального проявления болезни. В эти же периоды проявления болезни наблюдаются увеличение содержания окисленного глутатиона в плазме крови животных и снижение тиолдисульфидного отношения. Нарушения окислительно-восстановительного состояния тиолдисульфидной системы в плазме крови у БЭ-животных сохраняются с развитием болезни. Фактически концентрации GSH и его окисленной формы – глутатион дисульфида в основном определяют окислительно-восстановительное состояние клетки. Текучесть мембран прибелковых и липидных областей мембран эритроцитов у бульбэктомированных животных выше, чем у ложнооперированных практически во все сроки после операции. Следовательно, изменения структурного состояния мембраны и нарушение регуляции окислительно-восстановительного гомеостаза глутатиона у животных можно рассматривать как следствия не только окислительного стресса, характерного для поздней стадии болезни Альцгеймера, но и редуктивного (упрощенного) стресса в период до проявления болезни, который увеличивает риск дальнейшего развития деменции альцгеймеровского типа.
Список литературы
Бобкова Н.В., Нестерова И.В., Нестеров В.В. // Бюл. эксперим. биологии и медицины. 2001. Т. 131. № 5. С. 507.
Бобкова Н.В., Нестерова И.В., Александрова И.Ю. // Психиатрия. 2008. № 4–6. С. 34.
Porquet D., Andréz-Benito P., Griñán-Ferré C. et al. // Age. 2015. V. 37. P. 9747.
Lin M.T., Beal M.F. // Nature. 2006. V. 443. P. 787.
Wilkins H.M., Carl S.M., Weber S.G. et al. // J. Alzheimers Dis. 2015. V. 45. P. 305.
Riechter C., Gogvadze V., Laffranchi R. et al. // Biochim. Biophys. Acta. 1995. V. 1271. P. 67.
Владимиров Ю.Ф., Арчаков А.И. Перекисное окисление липидов в биологических мембранах. М.: Наука, 1972.
Герасимов Н.Ю., Голощапов А.Н., Бурлакова Е.Б. // Хим. физика. 2009. Т. 28. № 7. С. 82.
Molochkina E.M., Gerasimov N.Yu., Goncharova I.N. et al. // Chem. Phys. Lipids. 2008. V. 143. P. 94.
Floyd R.A. // Proc. Soc. Exp. Biol. Med. 1999. V. 222. P. 236.
Ziegler D.M. // Annu. Rev. Biochem. 1985. V. 54. P. 305.
McBean G.J., Aslan M., Driffiths R.G., Torrã R.C. // Redox Biology. 2015. V. 5. P. 186.
Sing R.P., Shashvsat S., Kapur S. // J. Indian Acad. Clinical Med. 2004. V. 5. P. 218.
Pastore A., Federici G., Bertini E., Piemonte F. // Clin. Chim. Acta. 2003. V. 333. № 1. P. 19.
Valko M., Leibfritz D., Moncol J. // Intern. J. Biochem. Cell Biol. 2007. V. 39. P. 44.
Zhang C., Rodriguez C., Spaulding J. // J. Alzheimers Dis. 2012. V. 28. № 3. P. 655.
Marí M., Collel A., Morales A., Montfort C. // Antioxid. Redox Signal. 2010. V. 12 P. 1294.
Marí M., Morales A., Collel A. // Biochim. Biophys. Acta. 2012. V. 1830. № 5. P. 3317.
Bomhard E., Mohr U. // Exp. Pathol. 1989. V. 36. № 3. P. 129.
Иваненко Г.Ф., Бурлакова Е.Б. // Изв. АН. Сер. биол. 2005. № 1. С. 9.
Карпова С.Г., Милюшкина Э.Г., Люсова Л.Р. и др. // Хим. физика. 2018. Т. 37. № 3. С. 40.
Бинюков В.И., Борунова С.Ф., Гольдфельд М.Г. и др. // Биохимия. 1971. Т. 36. № 6. С. 1149.
Вассерман А.М., Бучаченко А.Л, Коварский А.Л., Нейман И.Б. // Высокомолекуляр. соединения. A. 1968. Т. 10. С. 1930.
Pradhan D., Weiser M., Lumley-Sapanski K. // Biochim. Biophys. Acta. 1990. V. 1023. P. 398.
Negre-Salaure A., Auge N., Auala V. et al. // Free Radical. Res. 2010. V. 44. P. 1125.
Zarkovic N., Cipak A., Jaganjac M. // J. Proteomics. 2013. V. 92. P. 239.
Braidy N., Zarka M., Welch J., Bridge W. // Curr. Alzheimer Res. 2015. V. 12. № 4. P. 298
Padurariu M., Ciobica A., Hriten L. et al. // Neurosci. Lett. 2010. V. 469. P. 6.
Ghezzi P. // Biochim. Biophys. Acta. 2013. V. 1830. № 5. P. 3265.
Liedhegner E.A.S., Gao Xing-Huang, Mieyal J.J. // Redox. Signal. 2012. V. 16. № 6. P. 543.
Ballatori N., Krance S.M., Notenboom S. et al. // Biol. Chem. 2009. V. 390. P. 191.
Kemp M., Go Y.M., Jones D.P. // Free Radical Biol. Med. 2008. V. 44. P. 921.
Zhang X., Min X., Li C., Benjamin L.J. et al. // Hypertension. 2010. V. 55. № 6. P. 1412.
Badía M.-C., Giraldo E., Dasí F. et al. // Free Radical Biol. Med. 2013. V. 63. P. 274.
Lloret A., Fuchsberger T., Giraldo E., Vina J. // Curr. Alzheimer Res. 2016. V. 13. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика