

Химическая физика, 2022, T. 41, № 4, стр. 72-80
Моделирование адсорбции водорода на кластерах AunNim, AunCum и CunNim, n + m = 13
Н. В. Дохликова 1, *, А. К. Гатин 1, С. Ю. Сарвадий 1, С. А. Озерин 1, Е. И. Руденко 1, М. В. Гришин 1, Б. Р. Шуб 1
1 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: dohlikovanv@gmail.com
Поступила в редакцию 21.05.2020
После доработки 14.05.2021
Принята к публикации 20.05.2021
- EDN: RPEJTQ
- DOI: 10.31857/S0207401X22040021
Аннотация
В работе представлены результаты квантовохимического моделирования адсорбции атома водорода на биметаллических наночастицах AuNi, AuCu и CuNi. Установлено, что изменения электронной структуры этих кластеров зависят как от трансформации атомной структуры кластера, так и переноса заряда. Влияние переноса заряда проявляется наиболее заметно в кластерах, состоящих из атомов со слабо различающейся постоянной решетки Cu и Ni. При сильных различиях этого параметра изменения адсорбционных свойств будут определяться в большей степени трансформацией атомной структуры.
ВВЕДЕНИЕ
Современные адсорбенты [1], защитные покрытия [2], катализаторы [3], сенсоры [4], элементы электронных схем и улучшенные аккумуляторы [5] невозможно представить без использования различных функциональных наноразмерных материалов. Поэтому проблема разработки и поиска новых, обладающих более высокими характеристиками структурированных на наноуровне систем, остается актуальной. Вследствие этого покрытия на основе биметаллических наночастиц на подложках различной природы являются перспективными материалами с управляемыми физическо-химическими свойствами [6–8]. Совокупность многочисленных особенностей атомного и электронного строения, характерных для структур такого типа, может привести к нелинейному и неаддитивному изменению параметров биметаллической системы, т.е. к синергетическому эффекту [9]. Таким образом, взаимодействие между компонентами биметаллической, нанесенной наноразмерной структуры приводит к тому, что такие системы могут иметь сложное внутреннее строение (т.е. атомную и электронную структуры) из-за чего многие вопросы о механизмах протекания на них химических процессов, в том числе адсорбции, десорбции и др., остаются открытыми. Термин “атомная и электронная структура наноразмерной системы” означает совокупность связанных положительных ионов и электронов, которая характеризуется длинами и углами связей между атомами, а также распределением электронов по уровням энергии.
При использовании численного моделирования с применением теории функционала плотности (ТФП) можно получить подробную информацию об изменениях атомной и электронной структур моделей наночастиц – кластеров из состава описанных выше систем и проводить вычисления параметров адсорбции на них атомов и молекул. Для вычисления изменений электронной структуры проводятся расчеты плотностей состояний таких систем. Однако только сопоставление полученных данных численного моделирования с экспериментальными результатами сканирующей туннельной микроскопии и спектроскопии (СТМ/СТС), проведенных на хорошо определенных биметаллических структурированных на наноуровне покрытиях, позволяет достичь более полного понимания протекающих поверхностных реакций.
Биметаллические кластеры являются достаточно широко исследуемыми объектами. В ряде работ показано, что внедрение гетероатомов в кластер металла оказывает сильное воздействие на его физико-химические свойства [10–12]. Например, добавление атома переходного металла в кластер золота [13] увеличивает энергетическую стабильность, т.е. энергию образования. При этом его адсорбционные и электронные свойства определяются особенностями взаимодействия внешних электронных орбиталей атомов золота и примеси, а именно степенью перекрытия d–d и s–d орбиталей атомов золота и переходного металла. Например, атомы Ni и Pt усиливают взаимодействие между атомами в кластере благодаря большему перекрыванию s-орбиталей по сравнению с атомами Pd. Степень гибридизации s–d-орбиталей и выраженности релятивистских эффектов также влияют на тенденцию к образованию планарных структур, которая наиболее сильна в кластерах Au–Pt и Au–Pd, что доказано в работе [14].
Исследование окисления СО на кластерах типа MAu6 (M = Ni, Pt, Pd) [15] показало, что при допировании атомами Ni кислород адсорбируется на кластере в атомарной форме. Похожее исследование окисления СO на кластерах AuCu [16] выявило снижение реакционного барьера при добавлении Cu. При этом, однако, ухудшается селективность за счет более сильного связывания СО с кластером AuCu. Исследование устойчивости биметаллических кластеров NiCu на графене с дефектом (вакансией атома углерода) показало, что взаимодействие кластера с графеном через Ni энергетически выгодно за счет большего переноса заряда от Cu к Ni [17]. Применение ТФП для моделирования адсорбции CO2 на кластерах Pd–Cu показало, что икосаэдрический кластер Pd5Cu8 относится к числу “магических”, т.е. обладает повышенной стабильностью, а перенос заряда от Cu к Pd может снизить отравление CO2 [18]. Исследование каталитической реакции полугидрирования ацетилена C2H2 + H2 → C2H4 на биметаллических кластерах Pt–Cu показало, что происходит насыщение платины электронами, которое допускает образование комплекса этилена через π-связь [19].
На основании вышесказанного можно заключить, что биметаллические наночастицы представляют собой системы со сложно организованной внутренней структурой, физико-химические свойства которой обусловлены взаимным расположением и типом атомов, из которых она состоит. Поиск и исследование ключевых факторов, определяющих каталитические свойства наночастиц биметаллов, является одной из главных целей как настоящей работы, так и катализа на наночастицах в целом.
Ранее нами проведено исследование трансформации, т.е. изменения атомной и электронной структур кластеров Au [20], Cu и AuCu [21], Ni и AuNi [22], в том числе при взаимодействии с атомарным водородом. Цель нашей работы – определение качественных тенденций изменения энергии адсорбции атома водорода при варьировании элементного состава биметаллических кластеров AuCu, AuNi и CuNi и выявление взаимосвязи между элементным составом кластера, его электронной структурой и параметрами адсорбции атома водорода.
МЕТОДИКА ИССЛЕДОВАНИЙ
Численный эксперимент, моделирующий взаимодействие водорода с наночастицами на основе Au, Cu и Ni, проведен в рамках теории функционала плотности в обобщенном градиентном приближении и с функционалом PBE. Для расчетов использовали программные пакеты Quantum Espresso 5.1.1 (QE) [23] и OpenMX 3.8 (OMX) [24]. Релятивистские поправки движения электронов учтены при генерации ультрамягкого псевдопотенциала. Энергия отсечки базисного набора в пакете QE взята из рекомендаций к программным пакетам. Объем расчетной ячейки выбран после тестовых расчетов. Наборы примитивных базисных функций в пакете OMX взяты из документации к программному пакету. Спиновая поляризация не учитывалась. В результате моделирования определяли значения энергии и длины связи для каждого места адсорбции атома водорода на биметаллическом кластере, а также спроектированные плотности состояний отдельных атомов из ближайшего окружения адсорбата до и после взаимодействия с атомом водорода. Для получения более подробной информации кривые плотности состояний d- и s-орбиталей построены отдельно, поскольку при адсорбции водорода на наноразмерных системах переходных и благородных металлов они могут изменяться различным образом.
В качестве моделей наночастиц использованы 13-атомные кластеры. Ранее нами показано, что для качественного описания взаимодействия водорода с наночастицами достаточно кластеров именно такого размера [20]. Для исследований выбраны биметаллические кластеры двух различных типов: янус-кластеры и кластеры с одним замещенным атомом. Таким образом, в данной работе исследован ряд из шести кластеров с замещенным поверхностным атомом, обозначенных как: Au12Ni1, Cu12Ni1, Cu12Au1, Ni12Au1, Au12Cu1, Ni12Cu1, и шесть янус-кластеров, обозначенных как, Au7Cu6, Cu7Au6, Au7Ni6, Ni7Au6, Cu7Ni6, Ni7Cu6. Атомная структура всех биметаллических кластеров была оптимизирована, т.е. соответствовала локальному минимуму энергии. Для янус-кластеров Au7Cu6 и Au7Ni6 можно отметить тенденцию к планарности, свойственную небольшим кластерам золота [25].
Известно, что для кластеров металлов с одним и тем же количеством атомов характерно существование стабильных изомеров с близкими энергиями образования. Адсорбция атома водорода также может послужить причиной перехода от одного изомера к другому. Таким образом, изменения плотности состояний при адсорбции водорода могут быть связаны не только с образованием связи Н–Ме (Ме = Au, Ni, Cu), но и с трансформацией атомной структуры кластера в целом. Поскольку основное внимание в данной работе посвящено исследованию изменений спектров электронов биметаллических кластеров при взаимодействии с водородом и сопоставлению полученных результатов с экспериментальными данными, происходящая при адсорбции трансформация атомной структуры внесет значительные возмущения в рассчитываемые плотности состояний. Для того чтобы этого избежать, взаимное расположение атомов металла в кластере до и после взаимодействия с водородом остается неизменным. Далее в статье положение адсорбированного атома водорода на кластере будем называть сайтом и обозначать буквой “s”.
Для кластеров с внедренным атомом проведены исследования двух сайтов адсорбции: на примесном атоме – s1 и соседнем с ним атоме – s2. Для янус-кластеров проведены исследования четырех сайтов адсорбции: два на интерфейсе – si и два на максимальном удалении от интерфейса, на “вершинах” – sv. Ниже подсистема сайта указывается в скобках, например: сайт на вершине меди – sv(Cu), на интерфейсе золота – si(Au), на внедренном атоме никеля – s1(Ni).
КВАНТОВОХИМИЧЕСКОЕ МОДЕЛИРОВАНИЕ
На рис. 1 приведены кривые рассчитанных спроектированных плотностей d- и s-орбиталей и диаграммы энергетических уровней атомов гомогенных кластеров Au13, Cu13 и Ni13 при адсорбции водорода. Согласно полученным данным атом водорода взаимодействует с кластерами золота и меди со сдвигом плотности состояний d-орбиталей вниз относительно уровня Ферми (EF), снижением плотности состояний s-орбиталей и образованием разрыхляющего состояния (рис. 1а, б). При взаимодействии с никелем такого не происходит, поскольку состояния вблизи уровня Ферми образованы в основном d-орбиталями (рис. 1в). Значения энергии связи для сайтов с разным числом центров S представлены в табл. 1. Как видно из таблицы, центр распределения плотности состояний d-орбиталей атомов Au и Cu расположены ниже уровня Ферми, а у атома Ni он практически лежит на уровне Ферми. Можно отметить, что для более легких атомов меди и никеля предпочтителен трехцентровый сайт адсорбции, в то время как для золота наиболее энергетически выгоден одноцентровый сайт.
Рис. 1.
Спроектированные плотности состояний d- и s-орбиталей атомов кластеров металлов при адсорбции атома H и конфигурационные схемы адсорбционных комплексов: а – Au13, б – Cu13, в – Ni13. На всех графиках: сплошные линии – плотность состояний d-орбиталей, штриховые линии – плотность состояний s-орбиталей; 1 (черная кривая) – плотность состояний до адсорбции атома H, 2 (серая кривая) – плотность состояний после адсорбции атома H, σss/σsd – связывающая орбиталь, ${{\sigma _{{ss}}^{*}} \mathord{\left/ {\vphantom {{\sigma _{{ss}}^{*}} {\sigma _{{sd}}^{*}}}} \right. \kern-0em} {\sigma _{{sd}}^{*}}}$ – разрыхляющая орбиталь полученного адсорбционного комплекса атома Н и кластера.
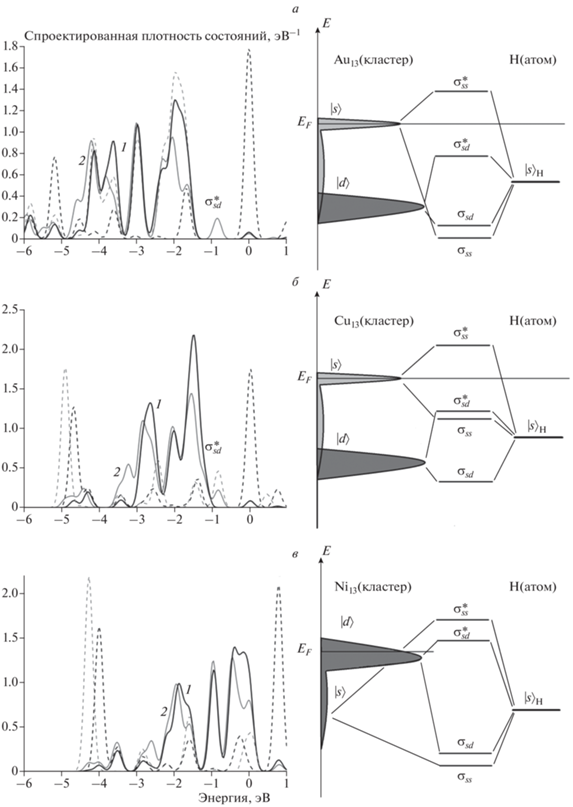
Таблица 1.
Энергия взаимодействия Eb и длина связи Rb атома H, адсорбированного на сайтах с различным числом центров S на поверхности монометаллических кластеров Au13, Cu13, Ni13; dz – центр распределения плотности состояний d-орбиталей
| Кластер | S | Eb, эВ | Rb, Å | dz, эВ |
|---|---|---|---|---|
| Au13 | 1 | –2.64 | 1.61 | –2.66 |
| 2 | –2.49 | 1.80 | ||
| 3 | –2.34 | 1.91 | ||
| Cu13 | 1 | –2.29 | 1.52 | –0.66 |
| 2 | –2.58 | 1.69 | ||
| 3 | –2.71 | 1.79 | ||
| Ni13 | 1 | –2.59 | 1.52 | 0.0038 |
| 2 | –3.014 | 1.67 | ||
| 3 | –3.098 | 1.77 |
Согласно предположению Норскова и Хаммера [26], существует взаимосвязь между химической активностью кластера и положением “центра тяжести” заполненных состояний его d-зоны. При расчете плотности состояний в биметаллических кластерах необходимо учитывать искажение атомной структуры, вызванное присутствием в них атомов разных типов. Биметаллический кластер может испытывать растяжение или сжатие в зависимости от атомного состава. При растяжении перекрытие атомных оболочек уменьшается, что приводит к сужению и сдвигу вверх распределения плотностей состояний относительно плотности состояний невозмущенного кластера. При сжатии перекрытие атомных оболочек, напротив, увеличивается, что приводит к расширению и сдвигу вниз распределения плотностей состояний.
Однако помимо трансформации атомной структуры в биметаллическом кластере возможно также перераспределение электронной плотности, что может привести к избыточному заряду на некоторых атомах. Избыток или недостаток электронной плотности может повлиять на взаимодействие с атомом водорода неоднозначно, так как приводит к изменению уровня Ферми и трансформации атомной структуры. Анализ заселенности по Малликену показал, что в биметаллических кластерах атомы золота всегда заряжены отрицательно (Au–), а атомы никеля – положительно (Ni+). Атом меди приобретает отрицательный заряд в окружении атомов никеля (Cu–) и положительный заряд – в окружении атомов золота (Cu+). Наибольший заряд приобретает единичный внедренный атом (табл. 2).
Таблица 2.
Расчетные значения зарядов на внедренных атомах Au, Cu, Ni в кластерах Au13, Cu13 и Ni13; e – заряд электрона
| Атом\кластер | Au13 | Cu13 | Ni13 |
|---|---|---|---|
| Au | 0 | –0.44e | –0.25e |
| Cu | +0.36e | 0 | –0.13e |
| Ni | +0.48e | +0.23e | 0 |
На рис. 2 приведены кривые, иллюстрирующие рассчитанные спроектированные плотности состояний d- и s-орбиталей биметаллических кластеров Cu7Au6, Ni7Au6, Cu7Ni6. На рисунке показаны результаты адсорбции водорода на сайтах si трех кластеров из шести исследованных, поскольку качественные тенденции к изменению электронной структуры кластеров Au7Cu6 и Cu7Au6, Au7Ni6 и Ni7Au6, Cu7Ni6 и Ni7Cu6, соответственно, совпадают. В табл. 3 представлены величины изменения энергии связей сайтов адсорбции атома водорода на поверхности биметаллических кластеров и сайтов адсорбции с тем же числом центров атома водорода на поверхности монометаллического кластера. Согласно резонансной модели хемосорбции расширение распределения плотности состояний будет приводить к уменьшению энергии связи, а сжатие, напротив, – к ее увеличению [27].
Рис. 2.
Спроектированные плотности состояний d- и s-орбиталей атомов кластеров AuCu, AuNi и CuNi при адсорбции атома Н для различных сайтов: а – сайт si(Au) кластера Cu7Au6, б – сайт si(Cu) кластера Cu7Au6, в – сайт si(Au) кластера Ni7Au6, г – сайт si(Ni) кластера Ni7Au6, д – сайт si(Cu) кластера Cu7Ni6, е – сайт si(Ni) кластера Cu7Ni6. На всех графиках: сплошные линии – до взаимодействия с атомом Н, штриховые – после взаимодействия с атомом Н; 1 (черная кривая) – плотность состояний d-орбиталей, 2 (серая кривая) – плотность состояний s-орбиталей. На вставках – атомные структуры кластеров биметаллов.
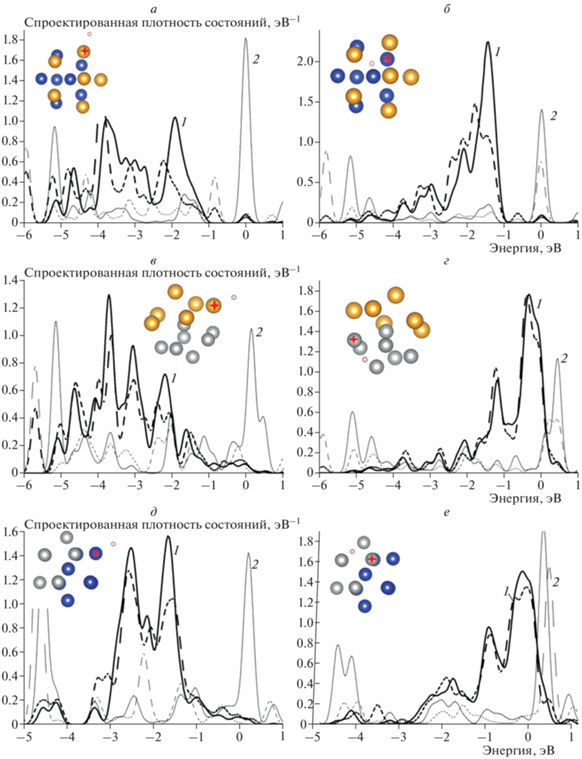
Таблица 3.
Разность между энергией связи сайта адсорбции атома водорода на поверхности биметаллических кластеров AuCu, AuNi, CuNi и энергией связи сайта адсорбции с тем же числом центров атома водорода на поверхности монометаллического кластера, ∆Eb
| Кластер | Сайт | ∆Eb, эВ | Кластер | Сайт | ∆Eb, эВ | Кластер | Сайт | ∆Eb, эВ |
|---|---|---|---|---|---|---|---|---|
| Au7Cu6 | si(Au) | 0.078 | Au7Ni6 | si(Au) | 0.051 | Cu7Ni6 | si(Cu) | –0.134 |
| sv(Au) | 0.068 | sv(Au) | 0.021 | sv(Cu) | –0.153 | |||
| si(Cu) | –0.146 | si(Ni) | 0.004 | si(Ni) | 0.044 | |||
| sv(Cu) | –0.176 | sv(Ni) | –0.027 | sv(Ni) | 0.071 | |||
| Cu7Au6 | si(Au) | 0.063 | Ni7Au6 | si(Au) | 0.038 | Ni7Cu6 | si(Cu) | –0.145 |
| sv(Au) | 0.075 | sv(Au) | 0.056 | sv(Cu) | –0.161 | |||
| si(Cu) | –0.178 | si(Ni) | –0.021 | si(Ni) | 0.055 | |||
| sv(Cu) | –0.021 | sv(Ni) | 0.013 | sv(Ni) | 0.067 |
Расчет плотностей состояний показал, что изменения энергетических спектров в янус-кластерах AuCu и AuNi заключаются в расширении и сдвиге вниз по энергии плотности состояний атомов золота (рис. 2а, в). Распределение плотности состояний атомов меди, напротив, сужается и сдвигается вверх по энергии (рис. 2б). Плотность состояний атомов никеля практически не меняется (рис. 2г) по сравнению с этими же параметрами в соответствующих монометаллических кластерах. Очевидно, что поскольку равновесное расстояние между атомами Au больше, чем между атомами Cu и Ni, основная тенденция к изменению плотности состояний обусловлена трансформацией атомной структуры и заметней проявляется для атомов с заполненной d-оболочкой, Au и Cu. В янус-кластере CuNi плотность состояний атомов Cu и Ni не меняется существенно по сравнению с плотностями состояний в монометаллических Cu- и Ni-кластерах, поскольку равновесные расстояния между атомами Cu и Ni отличаются не сильно (рис. 2д, е).
Указанные тенденции к изменению плотности состояний в целом коррелируют с изменениями энергии связей сайтов адсорбции атома Н на поверхности биметаллических кластеров и сайтов адсорбции с тем же числом центров атома Н на поверхности монометаллического кластера, представленными в табл. 3. Согласно результатам расчетов, энергия связи атома водорода на подсистеме Au уменьшается, а на подсистемах Cu и Ni увеличивается по сравнению с монометаллическими кластерами, что коррелирует с изменениями энергетических спектров под влиянием трансформации атомной структуры. В янус-кластере CuNi энергия связи атома Н на медной подсистеме увеличивается, а на никелевой – уменьшается. Однако плотность состояний при этом практически неизменна. Видимо, в таком биметаллическом кластере с близкими значениями равновесных расстояний между атомами большую роль будет играть перераспределение электронной плотности. Действительно, согласно данным табл. 2, подсистема Cu приобретает отрицательный заряд, а подсистема Ni – положительный, что согласуется с изменениями энергии связей.
При адсорбции водорода плотность состояний в янус-кластерах AuCu и AuNi качественно изменяется так же, как и в монометаллических кластерах. Происходит сдвиг плотности состояний d-орбиталей вниз по энергии и снижение плотности состояний s-орбиталей в околофермиевской области для атомов золото (рис. 2а, в) и меди в кластере AuCu (рис. 2б), при этом энергетический спектр атомов никеля не меняется (рис. 2г). В янус-кластере CuNi плотность состояний атомов Ni (рис. 2е) и атомов Cu в кластере CuNi (рис. 2д) практически не меняется. Полученные результаты моделирования коррелируют с результатами СТМ/СТС-экспериментов [20–22, 28]. Согласно им, после экспозиции в водороде наблюдалось снижение проводимости туннельного контакта для подсистемы Au в биметаллическом покрытии AuNi, причем вольт-амперные характеристики подсистемы Ni не менялись. При экспозиции в водороде биметаллического покрытия AuCu происходило снижение проводимости подсистем и золота и меди. На проводимость структуры CuNi экспозиция в водороде не оказывала заметного влияния.
На рис. 3 приведены кривые рассчитанных спроектированных плотностей состояний d- и s-орбиталей биметаллических кластеров Au12Ni1, Cu12Ni1, Ni12Au1, Cu12Au1, Au12Cu1, Ni12Cu1. Для краткости показаны результаты адсорбции атома Н на сайтах s1, поскольку изменения энергетических спектров на сайте s2 практически аналогичны изменениям в соответствующих монометаллических кластерах. Ниже представлены изменения энергии связей сайтов адсорбции атома водорода на поверхности биметаллических кластеров и сайтов адсорбции с тем же числом центров атома водорода на поверхности монометаллического кластера:
| Кластер | Au12Ni1 | Cu12Ni1 | Cu12Au1 | Ni12Au1 | Au12Cu1 | Ni12Cu1 |
| ∆Eb, эВ | 0.170 | 0.069 | 0.079 | 0.013 | –0.024 | –0.137 |
Рис. 3.
Спроектированные плотности состояний d- и s-орбиталей атомов кластеров Au12Ni1, Cu12Ni1, Ni12Au1, Cu12Au1 Au12Cu1, Ni12Cu1 при адсорбции атома Н для различных сайтов: а – сайт s1 кластера Au12Ni1, б – сайт s1 кластера Cu12Ni1, в – сайт s1 кластера Ni12Au1, г – сайт s1 кластера Cu12Au1, д – сайт s1 кластера Au12Cu1, е – сайт s1 кластера Ni12Cu1. На всех графиках: сплошные линии – до взаимодействия с атомом Н, штриховые – после взаимодействия с атомом Н; 1 (черная кривая) – плотность состояний d-орбиталей, 2 (серая кривая) – плотность состояний s-орбиталей. На вставках – атомные структуры кластеров биметаллов.
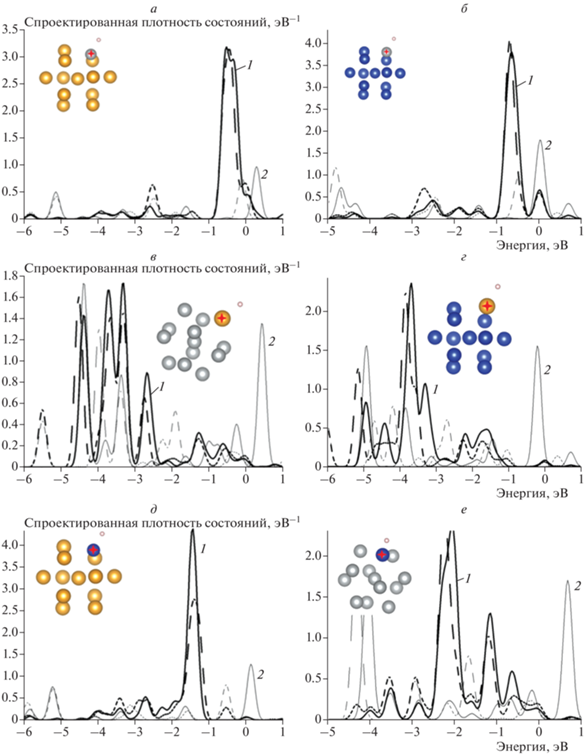
Поскольку на единственном замещенном атоме в биметаллическом кластере накопление заряда больше, можно предположить отклонения от резонансной модели хемосорбции.
Действительно, расчеты показали, что плотность состояний атома Ni+ сдвинута вниз (рис. 3а, б) и энергия связи с атомом Н меньше, чем в монометаллическом кластере. Как можно заключить, здесь проявляется влияние заряда, при доминирующей трансформации атомной структуры плотность состояний должна была сдвигаться вверх по энергии. Плотность состояний внедренного атома Au– сдвинута вниз по энергии (рис. 3в, г) и энергия связи с атомом Н меньше, чем в монометаллическом кластере, что говорит о преобладающем по-прежнему влиянии трансформации атомной структуры. Энергия связи внедренного атома Cu с атомом Н всегда больше, чем в монометаллическом кластере, при этом плотность состояний атома Cu+ в окружении атомов Au сдвигается вверх по энергии (рис. 3д), что указывает на преобладающее влияние трансформации атомной структуры, а плотность состояний атома Cu– в окружении атомов Ni сдвигается вниз по энергии (рис. 3е), что может быть связано с влиянием заряда.
ВЫВОДЫ
Для определения взаимосвязи изменений атомной и электронной структур наночастиц биметаллов и их адсорбционных свойств в данной работе с помощью ТФП-моделирования исследовано взаимодействие атома водорода с кластерами биметаллов AuNi, AuCu, CuNi. Установлено, что:
1. Изменения электронной структуры кластеров биметаллов зависят как от трансформации атомной структуры кластера, так и от переноса заряда между атомами кластера.
2. Преобладающий фактор при изменении электронной структуры обусловлен элементным составом кластеров и разностью равновесных межатомных расстояний моноатомных кластеров.
3. В кластерах, содержащих атомы с большой разностью равновесных межатомных расстояний, а именно AuCu, AuNi, основное влияние оказывает трансформация атомной структуры.
4. В кластерах CuNi межатомные расстояния отличаются незначительно, поэтому изменения адсорбционных свойств, главным образом, связаны с переносом заряда между атомами кластера.
5. Результаты численного моделирования коррелируют с данными экспериментов по адсорбции атома водорода на биметаллических структурированных на наноуровне покрытиях AuNi, AuCu, CuNi, выполненными с помощью методик сканирующей туннельной микроскопии/спектроскопии.
Все расчеты выполнены с использованием ресурсов Межведомственного суперкомпьютерного центра РАН (МСЦ РАН).
Работа выполнена в рамках государственного задания (номер госрегистрации темы АААА-А20-120013190076-0), а также при финансовой поддержке Российским фондом фундаментальных исследований (грант № 18-03-00060).
Список литературы
Liu W.Z., Xu X.J., Wang Y.J. et al. // Chin. Sci. Bull. 2010. V. 55. № 4–5. P. 373.
Sharma R.P., Raut S.D., Jadhav V.V. et al. // Mater. Lett. 2019. V. 237. P. 165.
Ling X.Y., Yan R.X., Lo S. et al. // Nano Res. 2014. V. 7. № 1. P. 132.
Khan S.B., Rahman M.M., Akhtar K. et al. // Plos One. 2014. V. 9. № 10. P. 7.
Zhou G., Zhu J., Chen Y.J. et al. // Electrochim. Acta. 2014. V. 123. P. 450.
Свиридова Т.В., Антонова А.А., Бойков Е.В. и др. // Хим. физика. 2013. Т. 32. № 4. С. 29.
Матвеева В.Г., Степачёва А.А., Шиманская Е.И. и др. // Хим. физика. 2019. Т. 38. № 11. С. 77.
Билалов Т.Р., Гумеров Ф.М., Габитов Ф.Р. // Сверхкритич. флюиды. 2018. Т. 13. № 2. С. 40.
Gross A. // Top. Catal. 2006. V. 37. № 1. P. 29.
Яржемский В.Г., Дьяков Ю.А., Изотов А.Д. и др. // Журн. неорган. химии. 2019. Т. 64. № 10. С. 1057.
Яржемский В.Г., Казарян М.А., Дьяков Ю.А. и др. // Там же. 2017. Т. 62. № 1. С. 69.
Яржемский В.Г., Изотов А.Д., Казарян М.А. и др. // ДАН. 2015. Т. 462. № 1. С. 55.
Yuan D.W., Wang Y., Zeng Z. // J. Chem. Phys. 2005. V. 122. № 11. P. 11.
Ranjan P., Chakraborty V. // J. Nanopart. Res. 2020. V. 22. № 2. P. 11.
Nigam S., Sahoo S.K., Sarkar P. et al. // Chem. Phys. Lett. 2013. V. 584. P. 108.
Ma L., Laasonen K., Akola J. // J. Phys. Chem. C. 2017. V. 121. № 20. P. 10876.
Montejo-Alvaro F., Rojas-Chavez H., Herrera-Rivera R. et al. // Physica E. 2020. V. 118. P. 6.
Baraiya B.A., Mankad V., Jha P.K. // Spectrochim. Acta A. 2020. V. 229. P. 8.
Ayodele O.B., Cai R.S., Wang J.G. et al. // ACS Catal. 2020. V. 10. № 1. P. 451.
Дохликова Н.В., Колченко Н.Н., Гришин М.В. и др. // Рос. Нанотехнол. 2016. Т. 11. № 1-2. С. 17.
Гатин А.А., Гришин М.В., Дохликова С.В. и др. // Росс. Нанотехнол. 2018. Т. 13. № 9–10. С. 3.
Гришин М.В., Гатин А.К., Дохликова Н.В. и др. // Росс. Нанотехнол. 2016. Т. 11. № 11–12. С. 49.
Giannozzi P., Andreussi O., Brumme T. et al. // J. Phys. Condens. Matter. 2017. V. 29. № 46. P. 30.
Ozaki T., Kino H. // Phys. Rev. B. 2004. V. 69. № 19. P. 19.
Assadollahzadeh B., Schwerdtfeger P. // J. Chem. Phys. 2009. V. 131. № 6. P. 11.
Hammer B., Norskov J.K. // Adv. Catal. 2000. V. 45. P. 71.
Hammer B., Norskov J.K. // Surf. Sci. 1996. V. 359. № 1–3. P. 306.
Гришин М.В., Гатин А.К., Дохликова Н.В. и др. // Хим. физика. 2019. Т. 38. № 1. С. 3.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика