

Химическая физика, 2022, T. 41, № 6, стр. 35-54
Алифатические полиэфиры для биомедицинских целей: дизайн и кинетические закономерности деградации in vitro
Т. А. Иванова 1, 2, *, Е. Н. Голубева 1, 3
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
2 Первый Московский государственный медицинский университет им. И.М. Сеченова
Москва, Россия
3 Федеральный исследовательский центр химической физики им. Н.Н. Семёнова
Российской академии наук
Москва, Россия
* E-mail: Tatianaivanovamsu@gmail.com
Поступила в редакцию 06.12.2021
После доработки 10.01.2022
Принята к публикации 20.01.2022
- EDN: KXUOJH
- DOI: 10.31857/S0207401X2206005X
Аннотация
В работе представлен краткий обзор методов создания и диагностики материалов на основе алифатических полиэфиров, используемых для создания временных протезов и лекарственных форм пролонгированного действия. После обзора полимерных материалов, применяемых в биомедицине, обоснована особая роль алифатических полиэфиров в достижении поставленных целей. Описана необходимость импрегнации полимерных биомедицинских изделий биологически активными соединениями, а также основные подходы, применяемые для этого в настоящее время. Особое внимание уделено “зеленым” сверхкритическим флюидным технологиям формирования пористых скаффолдов с одновременной импрегнацией, позволяющим управлять строением, размером, связанностью пор, а также распределением допантов. Обсуждаются возможности спектроскопии электронного парамагнитного резонанса (метод спинового зонда), позволяющие получить уникальные данные, которые характеризуют строение полимерных матриксов и особенности их деградации с выделением парамагнитных добавок. Приведены современные представления о механизмах деградации алифатических полиэфиров и высвобождения из них биологически активных веществ. Обсуждаются математические подходы к моделированию качественно различных экспериментальных кривых высвобождения низкомолекулярных веществ из пленок поли(D,L-лактида) разной толщины.
1. ВВЕДЕНИЕ
Полимерные материалы широко используют для создания лекарственных форм пролонгированного действия и восстановления больных или травмированных тканей в организме человека. Биосовместимые полимеры применяют в качестве имплантов в виде швов, костных пластинок, связок, сосудистых трансплантатов, сердечных клапанов, интраокулярных линз, зубных имплантов и медицинских устройств, таких как кардиостимуляторы, биосенсоры и т. д. [1, 2]. Также они применяются при изготовлении каркасов для тканевой инженерии [3, 4]. Выбор материала определяется его химическим составом, молекулярной массой, растворимостью, формой и структурой, гидрофильностью/гидрофобностью, смазывающей способностью, поверхностной энергией, водопоглощением и механизмом эрозии. В качестве биоматериалов используют как природные, так и синтетические полимеры.
В следующем разделе приведены примеры полимеров медицинского назначения. Показано, что синтетические полиэфиры являются одними из наиболее перспективных для медицины биоразлагаемымых полимеров. В разд. 3 обоснована необходимость импрегнации полимерных медицинских изделий биологически активными веществами. Далее в разд. 4 рассмотрены основные методы формирования полимерных конструкций для биомедицины. Раздел 5 посвящен описанию возможностей спектроскопии электронного парамагнитного резонанса (метод спинового зонда) для характеристики таких конструкций. В разд. 6 и 7 обсуждаются основные механизмы деградации полимеров и высвобождения из них биологически активных веществ, а также методы математического моделирования этих процессов. В заключении (разд. 8) подчеркнута необходимость предсказания динамики деградации временных протезов и закономерностей высвобождения лекарств и других важных добавок из препаратов пролонгированного действия.
2. ПОЛИМЕРЫ МЕДИЦИНСКОГО НАЗНАЧЕНИЯ
Природные полимеры являются компонентами биологических систем и производятся микроорганизмами, растениями и животными. Эти полимеры можно классифицировать следующим образом: белки (шелк, коллаген, желатин, фибриноген, эластин, кератин, актин, миозин), полисахариды (целлюлоза, амилоза, декстран, хитин, хитозан, альгинат, гликозаминогликаны) и полинуклеотиды (ДНК, РНК). Существует множество вариантов их применения: лейкопластыри, абсорбенты, косметические средства, средства доставки лекарств и матриксы для изготовления временных протезов. Природные полимеры активно взаимодействуют с клетками, что приводит к лучшему росту тканей в месте нахождения импланта, обладают слабым воспалительным откликом и высокоорганизованной молекулярной структурой. Однако при использовании материалов природного происхождения существует возможность передачи человеку различных заболеваний [5]. Серьезным препятствием для применения природных полимеров является и то, что они недоступны в больших количествах, дороги, сложны в обработке [6].
Синтетические полимеры широко применяются в медицине благодаря возможности варьирования их свойств (пористость, время разложения, механические характеристики) для конкретных целей. Такие полимеры могут быть получены путем полимеризации и поликонденсации мономеров. Синтетические полимеры часто дешевле полимеров природного происхождения, не имеют иммунологических проблем по сравнению с природными белками, могут быть произведены в больших количествах и имеют длительные сроки хранения. Многие коммерчески доступные синтетические полимеры проявляют предсказуемые и воспроизводимые физические свойства, характеризуемые такими параметрами как предел прочности, модуль упругости и скорость разрушения [7, 8], которые во многом сопоставимы со свойствами биологических тканей. Основные области применения синтетических полимеров – системы адресной доставки лекарств, рассасывающиеся шовные материалы и устройства для ортопедической фиксации, а также биоразлагаемые стенты для лечения заболеваний сердечно-сосудистой системы. К классу синтетических биоразлагаемых полимеров принадлежат полиангидриды, полиортоэфиры, полифосфазены и полиэфиры [6].
Наиболее перспективными синтетическими биоразлагаемыми полимерами являются полиэфиры. Их широко используют в медицине ввиду образования в организме нетоксичных продуктов гидролиза, прежде всего молочной и гликолевой кислот. Эти природные метаболиты в конечном итоге превращаются в воду и углекислый газ под действием ферментов в цикле трикарбоновых кислот и выводятся через дыхательную систему. Основные представители этого класса – полилактиды (PLA), полигликолиды (PGA) и их сополимеры (PLGA) с различными соотношениями лактидных и гликолидных фрагментов. Их процентное соотношение определяет скорость деградации таких образцов [9]. Различные полиэфиры [10–13] используют для изготовления временных протезов (винтов, штифтов, стержней, сеток [14] и пористых матриксов), систем доставки лекарств [6, 15–17], а также терапевтических инструментов. В дальнейшем именно классу алифатических полиэфиров мы уделим основное внимание. В табл. 1 представлены наиболее существенные области применения этих полимеров.
Таблица 1.
Свойства и области применения биоразлагаемых полимерных материалов [10]
| Полимер | Степень кристалличности, % | Срок биодеградации, недели | Область применения | Литература |
|---|---|---|---|---|
| Полигликолид/ полиглактин | 45–55 | 6–12 | шовные якоря, ремонт мениска, медицинские приборы, доставка лекарств | [141–143] |
| Поли(L-лактид) | 37 | 12–18 | фиксация перелома, интерференционные винты, шовный материал якоря, ремонт мениска |
[75, 143, 144] |
| Поли(D,L-лактид) | аморфный | 11–15 | ортопедические импланты, доставка лекарств | [142, 143, 145] |
| Поли(D,L-лактид-ко-гликолид), 85/15 | аморфный | 5–6 | интерференционные винты, шовные якоря | [75, 143, 146, 147] |
| Поли(D,L-лактид-ко-гликолид), 75/25 | аморфный | 4–5 | пластины, сетка, шурупы, доставка лекарств | [142, 143, 145] |
| Поли(D,L-лактид-ко-гликолид), 50/50 | аморфный | 1–2 | ортопедические импланты, доставка лекарств | [145, 146] |
3. ИМПРЕГНАЦИЯ ПОЛИЭФИРОВ БИОЛОГИЧЕСКИ АКТИВНЫМИ СОЕДИНЕНИЯМИ
Полимеры, используемые для изготовления материалов медицинского назначения, позволяют получать структуры с хорошими механическими свойствами, но не могут полностью обеспечить адгезию, миграцию, пролиферацию и дифференцировку клеток. Для решения этой задачи матриксы функционализируют путем введения в них биологически активных веществ (БАВ). Эти соединения выполняют различные функции, основными из которых являются предотвращение воспалительных процессов и стимуляция роста тканей. Существует несколько классов БАВ – факторы роста, гормоны, витамины, противовоспалительные препараты (как стероидные, так и нестероидные) и др. [18]. Факторы роста – это соединения природного происхождения, которые продуцируются многими клетками. Факторы роста активируют специфические программы развития, контролирующие миграцию, дифференцировку и пролиферацию клеток. Как правило, действие факторов роста зависит от их концентрации и времени воздействия на систему, часто уже незначительные количества данных веществ могут вызвать заметный эффект [19]. Имплантация полимерных конструкций в живой организм может вызвать локальное воспаление вследствие иммунного отклика [20, 21]. Поэтому большое значение имеет допирование полимеров медицинского назначения стероидными и нестероидными противовоспалительными препаратами, антибиотиками. Соединения, используемые на практике для введения в материалы медицинского назначения, представлены в табл. 2. Высвобождение БАВ из полимера должно быть дозируемым и контролируемым. В настоящее время достижения в области доставки лекарств основаны на рациональном дизайне полимерных материалов, разрабатываемых для конкретной цели и содержащих определенные лекарственные вещества.
Таблица 2.
Примеры БАВ, используемых на практике для допирования материалов медицинского назначения
| Допант | Каркас | Конечный вид конструкции | Высвобождение из полимерной матрицы | Литература |
|---|---|---|---|---|
| Прогестерон | поли(L-молочная кислота) (PLLA) | пленка | in vitro | [106] |
| Лидокаин | сополимер молочной и гликолевой кислот (PLGA) | пленка | in vitro | [87] |
| Ибупрофен | полиметиметакрилат (PMMA) | субмикронные пленки (25–250 нм) | – | [148] |
| Гидрохлорид ципрофлоксацина | сополимер молочной и гликолевой кислот (PLGA) | матриксы, прессованные из микрокапсул | in vitro и in vivo | [149] |
| Дексаметазон | хитозан | пористый матрикс | in vitro | [150] |
| 5-Флуорацил | полимолочная кислота (PLA), полимолочно-гликолевая кислота | пористый матрикс | in vitro | [43] |
| Ибупрофен | полиметилметакрилат–полимолочная кислота (PMMA–PLA) | пористый матрикс | in vitro | [117] |
| Ибупрофен | поликапролактон (PCL) | пористый матрикс | – | [151] |
| Индометацин | полимолочная кислота (PLA), сополимер молочной и гликолевой кислот (PLGA) | пористый матрикс | – | [42] |
4. МЕТОДЫ ПОЛУЧЕНИЯ ПОЛИМЕРНЫХ КОНСТРУКЦИЙ
Существуют несколько широко используемых на практике методов получения полимерных конструкций: метод литья полимерных объектов из растворителя и выщелачивания добавок [22, 23], мембранное ламинирование [23], формование из расплава [24], спекание микросфер [24], лазерная микростереолитография [25], сверхкритические флюидные технологии, в частности, сверхкритическая жидкостная инверсия фазы [26] и сверхкритическое вспенивание и импрегнация матриксов [24, 26–38].
Метод литья полимерных объектов из растворителя и выщелачивания частиц заключается в диспергировании просеянных минеральных частиц (обычно водорастворимых, например, NaCl) в полимерном растворе с последующим удалением растворителя и выщелачиванием соли [22]. Главными недостатками метода являются использование высокотоксичных растворителей и ограничение в толщине изготавливаемых мембран [23].
В методе мембранного ламинирования используются мембраны, ранее приготовленные путем литья из растворителя и выщелачивания частиц. Мембраны соответствующей формы пропитывают растворителем, а затем укладывают в трехмерную сборку со связанной системой пор. Объемные свойства конечных трехмерных каркасов зависят от свойств мембран. Благодаря этому методу можно создавать трехмерные полимерные конструкции с точными анатомическими формами, поскольку можно использовать компьютерное моделирование для создания шаблонов с желаемой формой имплантата. Однако этот метод требует больших затрат времени вследствие использования только тонких мембран. Другой недостаток состоит в том, что при наложении мембран может образовываться ограниченное количество взаимосвязанных пор [23].
Формование полимерных конструкций из расплава проводят при температурах, превышающих температуру стеклования аморфных полимеров и температуры плавления полукристаллических или кристаллических полимеров. Количество подаваемой теплоты зависит от температуры обработки, удельной теплоемкости полимера с учетом режима нагрева и используемого формообразующего элемента (штампа или пресс-формы). Эта технология масштабируема, воспроизводима и рентабельна, что способствует ее применению для создания полимерных фиксирующих элементов в ортопедии. Тем не менее у этого подхода есть и серьезные ограничения. Так, формование пористых материалов, например, структур для регенеративной медицины из расплава, необходимо комбинировать с методами порообразования, такими как выщелачивание твердых частиц (путем удаления порообразующих частиц) или фазовое разделение (путем избирательного растворения полимера). Добавление этапа порообразования убирает преимущество подхода – отсутствие использования растворителей. Более того, могут возникать проблемы с качеством получаемой конструкции из-за плохой теплопроводности полимеров в сочетании с их ограниченной термостабильностью [24].
Формирование пористой трехмерной структуры можно реализовать путем спекания микросфер. Сплавленные микросферы позволяют изготавливать специально разработанные формы, включая биоактивные молекулы или клетки, в зависимости от метода спекания. Каркасам на основе микросфер присущи отличные механические свойства в сочетании с контролируемым высвобождением лекарств с многообещающими результатами in vivo для регенерации костей и хрящей. Производство микросфер для спекания их в пористую конструкцию обычно требует применения органических растворителей, например, широко применяемый метод эмульсионно-растворительной экстракции. Но существуют и технологии изготовления, применяемые без использования растворителей: метод термического спекания, метод спекания со сжатым CO2, селективное лазерное спекание [24].
Трехмерные гибридные структуры могут быть сформированы методом лазерной микростереолитографии (двухфотонная стереолитография). Метод основан на пространственном сшивании реактивных полимеров, инициированном нелинейным поглощением двух фотонов, испускаемых лазером с фемтосекундной длительностью импульса. Благодаря этому подходу возможно пространственное сшивание в объеме материала даже в случае слабого поглощения инфракрасного излучения матрицей. Изготовление структуры с высоким разрешением поверхности осуществимо благодаря квадратичной зависимости скорости полимеризации от интенсивности света [25].
Получение полимерных матриксов с применением сверхкритических флюидных технологий имеет ряд преимуществ по сравнению с другими методами. Рассмотрим особенности этого подхода подробнее.
4.1. Сверхкритические флюидные технологии для получения полимерных матриксов
Сверхкритические флюидные технологии основаны на использовании веществ в агрегатном состоянии, промежуточном между жидкостью и газом. По плотности и растворяющей способности сверхкритический флюид (СКФ) похож на жидкость, по вязкости и сжимаемости – близок к газам. Коэффициент диффузии в СКФ значительно выше по сравнению с веществом в жидком состоянии. Для перехода вещества в сверхкритическое состояние необходимо превысить критические значения температуры и давления; подобный переход возможен для многих веществ, если они не разлагаются при критической температуре [39].
Наиболее широкое (более 80% случаев) применение в качестве рабочей среды получил сверхкритический диоксид углерода (скСО2). Он обладает рядом достоинств по сравнению с другими соединениями: легко переходит в сверхкритическое состояние (Ткр = 31.3 °С, Ркр = 7.36 МПа), относительно инертен, нетоксичен, взрыво- и пожаробезопасен, недорог и является хорошим растворителем для многих органических соединений [40]. Кроме того, возможно его многократное использование путем рециркуляции.
В настоящее время сверхкритические флюидные технологии на основе скСО2 активно используют при производстве полимерных форм для фармацевтического и медицинского применения. Это позволяет получать материалы без использования токсичных органических растворителей [26]. Существует несколько подходов к получению полимерных материалов в виде мембран (пленок) и пористых 3D-структур, среди которых жидкостная инверсия фазы и сверхкритическое вспенивание.
4.2. Сверхкритическая жидкостная инверсия фазы
Инверсия фаз или иммерсионное осаждение – процесс, основанный на погружении смеси, состоящей из раствора полимера и биологически активного вещества в антирастворитель, что вызывает термодинамическую нестабильность и последующее осаждение богатой полимером фазы [26]. Расслоение и кристаллизация приводят к формированию пор и структурированию на наноуровне. Скорости этих процессов зависят от концентрации полимера и свойств растворителя. Сверхкритическая инверсия фаз с СКФ включает использование скCO2 в качестве антирастворителя в методе иммерсионного осаждения. В этом случае температура и давление являются основными параметрами, влияющими на свойства скCO2 и, следовательно, на морфологию и пористость получаемых структур. После разделения фаз необходима промывка системы скCO2 для удаления растворителя, что также может привести к экстракции скCO2-растворимых соединений, например, полимерных примесей или биологически активных веществ [26].
4.3. Сверхкритическое вспенивание и импрегнация матриксов биологически активными соединениями
Пористые 3D-конструкции также получают при помощи “вспенивания” полимера. Образование пористой структуры происходит на этапе сброса давления в стационарном сверхкритическом реакторе. Характеристики сверхкритического флюидного процесса, такие как давление, температура, время выдерживания полимера в условиях СКФ и время сброса давления, в значительной степени влияют на характеристики получаемых материалов. Существует значительное количество работ, посвященных изучению влияния параметров сверхкритического процесса на свойства пористых полимерных материалов. Рассмотрим влияние каждого из этих параметров на свойства получаемых структур из полиэфиров подробнее (в частности, полилактида, полилактоглиголида и поликапролактона).
Однозначно выявлено, что для полиэфиров при увеличении температуры в диапазоне 35–60 °C происходит формирование более крупных по размеру пор [24, 26, 31–33]. В некоторых работах при повышении температуры также наблюдали увеличение пористости для полилактогликолида (38–50 °C) и поликапролактона (>45 °C), а в работе [31] – образование более открытых пор для структур из полилактида в диапазоне температур 35–55 °C. Однако повышение температуры сопровождается образованием менее однородных структур для поликапролактона и полилактида в диапазоне 40–60 °C [24, 33]. В работе [34] для поликапролактона была предложена оптимальная рабочая температура около 45 °C. Это связано с тем, что ниже этой температуры растворимость CO2 в полимере невысокая и пористость получаемых структур недостаточная. При температурах выше 45 °C CO2 активнее проникает в полимер, что вызывает увеличение пористости, но приводит к получению неоднородной структуры, а также к проблемам с контролем размера пор.
В большинстве работ установлено, что при повышении давления в системе в диапазоне 6–23 МПа, средний размер пор в получаемых материалах из полилактида и полилактогликолида уменьшается [26, 31–33] (рис. 1а). Однако существуют работы, где отмечены другие тенденции. В работе [34] при повышении давления в диапазоне 12–16 МПа (45 °C, время выдерживания – 3 ч) для структур из поликапролактона средний размер пор сначала увеличивается (12–14 МПа), затем уменьшается (14–16 МПа), но в целом размер пор увеличивается (сравнение показателей для давлений 12 и 16 МПа). В работе [32] для полилактогликолида показано, что при увеличении давления в диапазоне 15–20 МПа размер пор уменьшается; затем при дальнейшем повышении давления до 30 МПа размер пор увеличивается, хотя для похожего на полилактоглиголид полилактида в работе [26] средний размер пор уменьшается при увеличении давления (10–23 МПа). Поэтому однозначно сказать, как связано изменение давления с изменением среднего размера пор нельзя. Влияние давления на размер пор зависит также и от других параметров сверхкритического процесса, таких как температура, время выдерживания и время сброса давления. Зависимость пористости матриксов от давления также неоднозначна: в работе [32] установлено, что пористость немного уменьшается при повышении давления в диапазоне 15–25 МПа, а при дальнейшем увеличении до 30 МПа она не меняется; в работе [34] показана противоположная тенденция – при повышении давления в диапазоне 12–16 МПа пористость немного увеличивается.
Рис. 1.
а – Зависимость размера пор от давления для скаффолдов из поли-D,L-лактида (параметры процесса вспенивания: T = 40 °С, время сброса давления – 12 мин); б – зависимость размера пор от времени сброса давления для скаффолдов из поликапролактона. Для вспенивания скаффолдов в качестве пластификатора был использован скCO2 [26, 33].
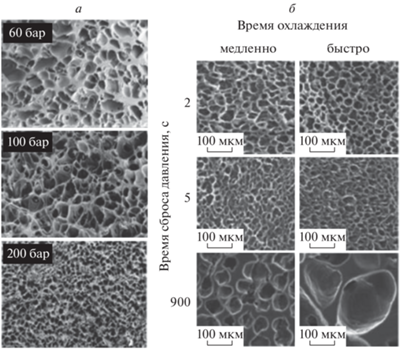
Многие эксперименты подтверждают гипотезу о том, что при увеличении времени выдерживания поликапролактона и полилактогликолида в сверхкритических условиях (0.5–5 ч) образуются более однородные структуры [26], средний размер пор уменьшается, а кривая распределения пор по размерам становится более узкой [24, 31, 35]. Например, при давлении 120 бар и времени выдерживания 1 ч получаются неоднородно вспененные матриксы, но если увеличить время выдерживания до 4 ч, то уже при давлении 10 МПа формируются однородно вспененные конструкции (температура выдерживания и время сброса давления одинаковые – 35 °C и 120 мин) [31]. Кроме того, в работе [35] структуры, выдержанные в сверхкритических условиях в течение 5 ч, были охарактеризованы кривой с узким распределением пор по размерам со значениями, находящимися в диапазоне 1–500 мкм, удовлетворяющими критериям применимости конструкций для инженерии костной ткани. Параметры сверхкритического флюидного процесса составляли: Т = 39 °C, p = 14 МПа, скорость сброса давления составляла = 1.8 г CO2/мин.
Большое влияние на структуру материала оказывает скорость сброса давления [41], поскольку именно в процессе выхода диоксида углерода происходит вспенивание полимера. Согласно данным работ [31, 33, 36, 37], при уменьшении скорости сброса поры получаются более взаимосвязанными, пористость структур увеличивается [32, 37]. Принято считать, что при увеличении времени сброса давления повышается однородность образца [26, 42], а анизотропия пор уменьшается [33]. В работе [31] показано, что при небольших временах сброса (2 и 5 мин) происходит формирование неоднородных структур. Однако во многих работах утверждается, что при увеличении времени сброса давления для полимерных конструкций из полилактогликолида и поликапролактона (0–120 мин) происходит увеличение среднего размера пор [26, 31–33, 36, 37] (рис. 1б). Напротив, в работе [42] авторы наблюдали противоположную зависимость – уменьшение размера пор в получаемой структуре при увеличении времени сброса давления. В целом можно заключить, что имеющиеся в литературе данные о влиянии параметров сверхкритического процесса на свойства пористых материалов недостаточны и во многом противоречивы.
Для предотвращения воспалительных процессов и стимуляции роста тканей в полимерные конструкции возможно введение биологически активных веществ. Процесс импрегнации может быть осуществлен одновременно с процессом создания пористой структуры [27–29, 38, 43]. В этом случае молекулы лекарственного вещества проникают в полимерную матрицу из раствора в сверхкритическом флюиде. После возвращения системы к нормальным условиям и испарения флюида вводимое соединение остается внутри полученной полимерной конструкции [44].
В работе [30] оценено влияние сразу нескольких параметров импрегнации полимеров в скСО2 на характеристики получаемых вспененных матриксов. Среди других работ она выделяется тем, что в ней рассматривали равномерность распределения вводимого соединения по матрице. В качестве полимера использовали поли(D,L-лактид), в качестве допанта – нитроксильный радикал TEMPOL, а изучали образующиеся структуры с помощью спектроскопии электронного парамагнитного резонанса (ЭПР). Структуры этого и других упоминаемых в обзоре нитроксильных радикалов приведены на рис. 2.

Авторами показано, что равномерность распределения допанта по матрице зависит от времени выдерживания в сверхкритических условиях, давления в системе и времени сброса давления. При давлении ниже 16 МПа были получены неравномерно вспененные образцы. Равномерность распределения вводимого соединения возрастает при увеличении времени выдерживания в сверхкритических условиях с 20 до 480 мин и снижении давления в системе с 22 до 16 МПа. Показано, что чем медленнее осуществляется спуск давления, тем более равномерно спиновый зонд распределяется во вспененном полимере. Авторы работы также рассмотрели влияние скорости сброса давления на такую важную характеристику трехмерных матриц как степень проницаемости пор. Для этого вспененные полимерные конструкции выдерживали в течение суток в чернилах для принтера MOORIM (Южная Корея), затем сушили в течение двух дней в эксикаторе. Оценка прокрашенности внутренней части образцов свидетельствовала о проницаемости пор для внешней среды. Оказалось, что взаимосвязанные поры можно получить при сбросе давления в течение 20 мин и более.
С целью увеличения механической прочности матриксов в них могут быть добавлены неорганические включения. Так, например, совместное введение гидроксиапатита и коллагена способствует увеличению механической прочности конструкций [26]. Существуют методики, при которых за сверхкритическим пенообразованием следует выщелачивание, что обеспечивает высокую пористость полученных конструкций, однако в этом случае существует риск выделения биологически активных допантов во время выщелачивания [26]. Уникальные данные, характеризующие строение полимерных матриксов и их деградацию с выделением парамагнитных БАВ, можно получить с использованием спектроскопии электронного парамагнитного резонанса.
5. СПЕКТРОСКОПИЯ ЭЛЕКТРОННОГО ПАРАМАГНИТНОГО РЕЗОНАНСА (МЕТОД СПИНОВОГО ЗОНДА)
Спектроскопия электронного парамагнитного резонанса (ЭПР) – метод, применимый для диагностики систем с ненулевым суммарным электронным спиновым моментом. Так как большинство материалов являются диамагнитными, то для исследований удобно применять метод спиновых зондов и меток. В первом случае к исследуемой системе добавляют небольшое количество стабильного парамагнитного вещества (спинового зонда). Во втором – парамагнитный фрагмент “пришивают” к исследуемой молекуле химическими связями. В качестве спиновых зондов и меток, как правило, используют парамагнитные молекулы с большой анизотропией g-фактора и констант сверхтонкого взаимодействия (СТВ). Одними из наиболее широко используемых спиновых зондов являются стабильные нитроксильные радикалы [45–53], которые обладают значительной пространственной анизотропией константы СТВ неспаренного электрона с ядром атома азота 14N (IN = 1). За счет высокой анизотропии СТВ спектры ЭПР нитроксильных радикалов проявляют высокую чувствительность к ориентационной упорядоченности и вращательной подвижности парамагнитных частиц. В качестве количественной характеристики подвижности используют время вращательной корреляции – среднее время, необходимое для поворота частицы на 90°.
В спектрах жесткого предела, например, зонда в жестких полимерных матрицах, движение частиц заторможено (область медленных движений, время вращательной корреляции находится в пределах ‒10–9÷–10–7 с), частицы ориентированы случайным образом. В этом случае спектр ЭПР характеризуется сложной структурой и представляет собой сумму спектров парамагнитных частиц всех возможных ориентаций. Это связано с анизотропией g-фактора и константы СТВ. Относительная интенсивность компонент в спектре ЭПР, их ширина и положение зависят от подвижности радикалов, их взаимного расположения и физико-химических свойств окружения [54]. Поскольку степень усреднения магнитных параметров радикалов зависит от их вращательной подвижности, то форма спектров ЭПР парамагнитных зондов в полимерах отражает подвижность зондов, которая в свою очередь связана с подвижностью полимерных цепей, а также со свободным объемом полимера. В менее вязких средах наблюдается большая подвижность зонда (время вращательной корреляции составляет 5⋅10–11 ÷ –10–9 с), сверхтонкое взаимодействие усредняется, что проявляется в сближении и сужении линий. В жидких невязких средах спектр ЭПР нитроксильных радикалов представляет собой узкий триплет, где расстояние между линиями равно среднему значению константы СТВ. При помещении полимера в жидкую среду молекулы жидкости диффундируют внутрь полимера, при этом часть радикалов из жесткого полимерного окружения переходит в менее вязкую жидкую среду, что приводит к появлению в спектре ЭПР узких компонент [48]. Моделирование спектров ЭПР путем специально разработанных программ [55, 56] позволяет оценивать времена вращательной корреляции зондов в разных средах [54].
Общее число парамагнитных молекул в образце пропорционально площади под кривой спектра поглощения электромагнитного излучения. Так как спектр ЭПР регистрируют в виде первой производной спектра поглощения, то для определения числа спинов в образце проводят двойное интегрирование экспериментального спектра. В отличие от спектрофотометрии в спектрах ЭПР, как правило, проявляются только разрешенные переходы. Поэтому для определения количества парамагнитных молекул достаточно сравнить интегральную интенсивность спектра изучаемого образца с интегральной интенсивностью спектра образца с известным количеством парамагнитного вещества (стандарта). В современных спектрометрах информация об интегральной интенсивности спектра стандарта сохраняется в памяти прибора. Таким образом, определение концентрации вещества методом ЭПР не включает в себя трудозатратную и вносящую значительную погрешность стадию определения вероятности изучаемого перехода (в спектрофотометрии – коэффициента экстинкции).
Данные, полученные в работе [57], позволяют утверждать, что при регистрации спектров с использованием современных ЭПР-спектрометров в сочетании с программным обеспечением для компьютерного анализа спектров вклад ошибки интегрирования становится менее существенным, чем считали раньше. При отношении сигнал/шум <20 вклад ошибки быстро растет с уменьшением этого параметра. Если это отношение больше 20, вклад ошибки интегрирования становится незначительным, и основной вклад в погрешность измерения концентрации вносят ошибки определения фактора добротности, обычно близкие к 8–10%. В среднем погрешность такого метода для определения количества нитроксильных радикалов в диапазоне 9.8 ⋅ 1014–5.6 ⋅ 1016 частиц в образце составляет 10–15%.
В том случае, когда отношение сигнал/шум в спектре ЭПР менее 20, ошибка определения количества парамагнитных частиц путем двойного интегрирования спектра ЭПР резко возрастает. Для таких систем недавно разработан количественный метод, основанный на конволюции экспериментальных зашумленных спектров со спектром ЭПР аналогичных частиц при больших концентрациях [58].
Cпин-обменные и диполь-дипольные взаимодействия влияют на форму линий спектров ЭПР [54]. Взаимодействия первого типа преобладают в невязких средах, вторые – могут проявляться в системах с заторможенным вращением, когда расстояния между парамагнитными частицами не превышают 30–40 Å. На этом основано определение локальной концентрации парамагнитных молекул (или, точнее, среднего расстояния между ними) в твердых образцах. Так, например, в работе [59] описана методика оценки средних расстояний между нитроксильными радикалами на основании анализа формы линий спектров ЭПР, зарегистрированных в отсутствие вращения парамагнитных молекул (в жестком пределе). Таким образом, с помощью спектроскопии ЭПР можно определять как среднюю, так и локальную концентрацию парамагнитных частиц в образце и установить наличие локального концентрирования спинов в полимерной матрице.
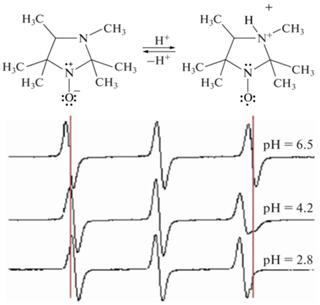
Существуют нитроксильные радикалы, спектры которых чувствительны к pH среды. С их помощью можно определить pH локального окружения парамагнитной молекулы путем измерения расстояния между линиями сверхтонкой структуры. Рассмотрим принцип рН-чувствительности нитроксильных радикалов на примере нитроксильных производных имидазолидина (рис. 3). Протонирование азота в положение “3” приводит к уменьшению спиновой плотности на атоме азота нитроксильного фрагмента, что отражается в уменьшении константы СТВ. При значениях pH существенно отличающихся от pKa, в системе присутствуют только протонированные или непротонированные нитроксильные радикалы. При значениях pH, близких к pKa, спектр ЭПР является суперпозицией спектров протонированной и непротонированной форм [46, 60].
Рис. 3.
Изменение расстояния между линиями сверхтонкой структуры в спектре ЭПР при протонировании нитроксильного производного имидазолидина [60].
ЭПР-томография позволяет генерировать изображения, показывающие пространственное распределение и свойства парамагнитного вещества по всему объему образца. В основе данного метода лежит идея использования градиента магнитного поля [60]. Так, например, ЭПР-томография позволяет “увидеть” диффузию воды внутрь полимерной матрицы, допированной парамагнитным соединением, при помещении ее в водную среду. В случае использования pH-чувствительных нитроксильных радикалов можно зафиксировать градиент pH в образце, который меняется с течением времени в процессе набухания полимерной матрицы [52, 61]. Современные приборы позволяют получать информацию с шагом около 10 мкм. Главным недостатком ЭПР-томографии является высокая стоимость оборудования, поэтому работ с использованием данной методики немного.
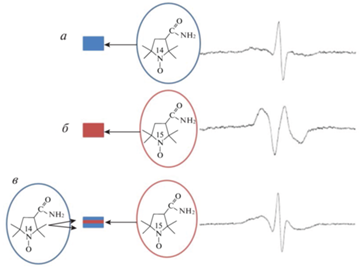
Вводя нитроксильные зонды, содержащие различные изотопы атомов азота, в разные области образца, можно получать данные о его строении без использования ЭПР-томографии. Спектры нитроксилов, содержащих 14N и 15N в составе нитроксильного фрагмента, демонстрируют разное число линий в спектре ЭПР (15N, спин ядра – 1/2, две линии в спектре; 14N, спин ядра – 1, три линии в спектре). Так, в работе [62] были сформированы “сэндвич”-подобные таблетки, содержащие 14N‑меченые молекулы нитроксила во внешних слоях и 15N-меченые молекулы нитроксила в центральном слое (рис. 4). При помещении таблеток в буферный раствор можно было увидеть последовательное набухание внешних и внутренних слоев образца.
Рис. 4.
Спектр ЭПР “сэндвич”-таблетки (в), представляющий собой суперпозицию спектров ЭПР 14N- (а) и 15N-меченых (б) молекул нитроксила [62].
В целом можно заключить, что спектроскопия ЭПР открывает широкие возможности исследования полимерных образцов, допированных парамагнитными веществами. Это – точный и неразрушающий метод, при помощи которого можно определять степень равномерности распределения парамагнитного допанта в полимерной матрице на макроскопическом и молекулярном уровне. Существенная зависимость формы спектра нитроксильного радикала от вращательной подвижности парамагнитной молекулы дает возможность характеризовать локальное окружение парамагнитного соединения; одновременно наблюдать за молекулами, находящимися в полимерной матрице и в заполненных жидкостью порах; изучать внутреннюю структуру, механизмы набухания полимерных матриц и кинетические закономерности высвобождения молекул допанта во внешнюю среду [46, 48, 61]. Другие, более известные методы, используемые для исследования процессов деградации полимерных матриксов и высвобождения их них биологически активных веществ и спиновых зондов, будут представлены при рассмотрении закономерностей этих процессов.
6. ДЕГРАДАЦИЯ ПОЛИМЕРОВ
Как говорилось ранее, применение полимерных материалов в медицине предполагает знание закономерностей набухания и разложения полимерных матриц в биологических средах, а также кинетических закономерностей высвобождения молекул допанта. Для создания временных протезов различных тканей и органов чаще всего используют полилактиды и полилактогликолиды. Такие полимеры разлагаются путем прямого гидролиза сложноэфирной связи в водной среде и дают легко усваиваемые, не причиняющие вреда организму, продукты разложения [63–65].
На разложение полимера влияет множество структурных, химических и технологических факторов [9]:
• природа полимера, молекулярный вес, молекулярно-массовое распределение;
• размеры и форма полимерного образца;
• морфология полимерной матрицы (степень кристалличности, наличие микроструктур, остаточные напряжения);
• природа и количество допанта;
• адсорбированные и поглощенные полимером соединения (вода, липиды, ионы, белки, кислород и т.д.);
• место имплантации.
Поли-D,L-лактид (PDLLA) представляет собой аморфный полимер, гидролитическая деградация которого в водном растворе происходит путем расщепления сложноэфирной связи. Этот процесс контролируется следующими основными параметрами: константой скорости реакции, количеством поглощенной воды, коэффициентами диффузии фрагментов цепей внутри набухшего полимера и растворимостью продуктов разложения [66]. Дополнительными параметрами, влияющими на гидролиз, являются молекулярная масса полимера [67], температура и pH раствора [67–70]. Было обнаружено, что в диапазоне Mn от 8 ⋅ 104 до 4 ⋅ 105 г/моль влияние молекулярной массы на скорость разложения PLLA незначительно, при значениях Mn ниже 4 ⋅ 104 г/моль гидролитическое разложение значительно ускоряется [71, 72]. Более низкой молекулярной массе соответствуют более высокая молекулярная подвижность, а также более высокая вероятность образования водорастворимых олигомеров и мономеров, что увеличивает скорость диффузии воды и, следовательно, ускоряет деградацию PDLLA [71]. В случае матриц на основе полилактогликолидов при увеличении содержания гликолида процесс деградации протекает быстрее, так как более высокое содержание гликолида делает полимер более гидрофильным, что способствует лучшему проникновению молекул воды между полимерными цепями [73].
Разложение полимерной матрицы может протекать по двум альтернативным механизмам: поверхностная (гетерогенная) и объемная (гомогенная) эрозия [74]. В случае поверхностного механизма разрушение полимера происходит намного быстрее, чем проникновение воды в объем, следовательно, разложение происходит главным образом во внешнем полимерном слое. При протекании процесса по объемному механизму матрица разлагается медленно, и поглощение системой воды происходит быстрее, чем разложение полимера. В этом случае полимер набухает, и полимерные цепи расщепляются по всему объему. Следует отметить, что, как правило, полимеры могут разрушаться как по гомогенному, так и по гетерогенному механизму в зависимости от условий протекания процесса и архитектоники образца [75].
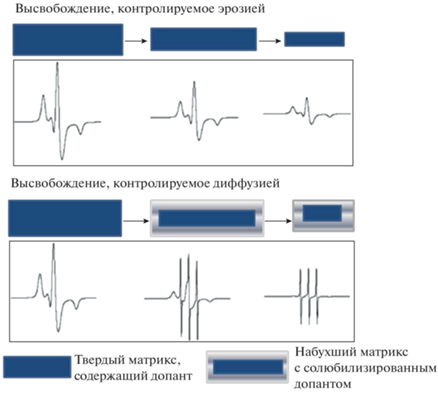
В работах научного коллектива под руководством проф. К. Мэдера метод спинового зонда был использован для установления закономерностей набухания полимерных матриц и высвобождения из них парамагнитных молекул во внешнюю среду [46, 48, 60, 62, 76]. Было показано, что разные механизмы высвобождения приводят к различным изменениям интенсивности и формы спектра ЭПР полимерной пленки (рис. 5). При высвобождении вещества в результате поверхностной эрозии матрицы интенсивность сигнала уменьшается, однако форма спектра остается неизменной в течение всего времени высвобождения. Если высвобождение вещества из матрицы сопровождается диффузией воды внутрь полимерного материала, меняются и интенсивность, и форма спектра ЭПР. В спектре появляются узкие компоненты подвижных (быстро вращающихся) радикалов, которые находятся в областях, заполненных жидкостью [60].
Рис. 5.
Изменения спектров ЭПР спиновых зондов при различных механизмах высвобождения парамагнитных молекул из полимерной матрицы [60].
В работе [77] представлена иллюстрация диффузии молекул воды внутрь полимерной матрицы – спектры ЭПР TEMPOL в пленке PDLLA (Mn = 7200), зарегистрированные при различных периодах набухания. Спектры набухших образцов представляют собой комбинацию широкого спектра ЭПР радикалов в “сухом” полимере и узкого триплетного сигнала, интенсивность которого со временем увеличивается. Этот сигнал представляет собой спектр радикалов, локализованных в областях полимера, в которые проникла вода. Анализ спектра ЭПР позволяет оценить соотношение подвижной и неподвижной форм радикала в набухшей матрице.
Количественно набухание полимерного материала характеризуют индексом (степенью) набухания SI, % = (Mt – M0)/M0 · 100 [78–80]. За изменениями молекулярно-массового распределения полимера (MWD), средневесовой молекулярной массы (Mw), среднечисловой молекулярной массы (Mn) в ходе гидролиза наблюдают при помощи гель-проникающей хроматографии [79, 81–87].
Важной особенностью гидролиза полиэфиров является его автокаталитический характер. Известно, что в кислой среде гидролиз полиэфиров протекает быстрее, чем в нейтральной. В ходе разложения вследствие образования молочной и гликолевой кислот – продуктов гидролиза – происходит закисление внутренних областей полимера, а также среды, в которой находится полимерный материал, что приводит к автокаталитическому ускорению процесса разложения [78, 88–90].
В настоящее время для определения pH внутри полимерной матрицы в качестве спиновых зондов широко используются pH-чувствительные радикалы [45–48, 60]. Так, в статье [47] измерено значение pH внутри биоразлагаемых микросфер PLA и PLGA во время эрозии. Большое число факторов влияет на значение pH внутри биоразлагаемого полимера на разных стадиях его набухания и деградации (степень набухания, присутствие олигомеров и мономеров, их кислотно-основные свойства, концентрация и кислотно-основные свойства лекарственного допанта и т.д.). При этом закисление матрицы может оказывать существенное влияние на стабильность, растворимость, биодоступность, фармакокинетику лекарственного средства и, в конечном итоге, на терапевтическую эффективность лекарственной формы [46].
Деградация матриц PLLA, характеризующихся определенной степенью кристалличности, происходит еще более сложным образом. Кристаллические области являются более устойчивыми к гидролизу по сравнению с аморфными областями, поскольку доступ молекул воды к цепям внутри жестких кристаллических областей сильно ограничен [91]. Гидролитическая деградация матриц из частично закристаллизованных полимеров протекает в две стадии. На первом этапе вода диффундирует в аморфные области, где начинается гидролиз сложноэфирных связей. Степень кристалличности образца в этом случае может даже увеличиваться. Вторая стадия начинается после деградации большей части аморфного участка. Гидролитическая атака происходит от края к центру кристаллических доменов [92].
Химические взаимодействия между молекулами допанта и полимером могут оказывать сильное влияние на кинетику разложения полимера и высвобождения лекарственного средства [81]. Лекарственные средства могут ускорять или замедлять скорость разложения полимера. В частности, гидрофобное лекарственное средство имеет тенденцию ограничивать поглощение воды и, таким образом, уменьшать скорость разложения, тогда как гидрофильное вещество оказывает противоположное влияние на поглощение воды и скорость разложения. Для лекарств, обладающих кислотными свойствами, можно ожидать более быстрого гидролиза сложноэфирных связей вследствие кислотного катализа. Напротив, в случае лекарственных средств с основными свойствами можно наблюдать два эффекта: основной катализ расщепления сложноэфирной связи и нейтрализацию карбоксильных концевых групп полимерных цепей, что минимизирует или устраняет автокаталитический эффект кислотных концов цепи. Например, деградация PDLLA в присутствии “основного” лекарственного средства, тиоридазина, который является третичным аминосоединением, ускоряется, что связано с нуклеофильной природой аминогруппы [78, 93].
Благодаря способности к восстановлению и окислению нитроксильных радикалов можно оценивать скорость проникновения восстановителей в полимерную матрицу [60]. Так, при использовании нитроксильных радикалов в качестве зондов для исследования биологических объектов ex vivo и in vivo их восстановление аскорбиновой кислотой и серосодержащими белками до диамагнитных гидроксиламинов является серьезной проблемой [94].
7. ВЫСВОБОЖДЕНИЕ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ И ЗОНДОВ ИЗ ПОЛИМЕРНЫХ МАТРИЦ
Процессы высвобождения введенных в полимерные матрицы веществ при их имплантации непосредственно связаны с процессами гидролиза, набухания и деградации полимерных материалов. Эксперименты по высвобождению БАВ из полимерных матриц in vivo моделируют при помощи экспериментов in vitro. Их проводят в средах, имитирующих биологические жидкости при температуре 37 °С, чаще всего в качестве среды используют нейтральные буферные растворы (рН ~ 7). Закономерности высвобождения определяются составом полимера, степенью его полимеризации, природой БАВ, распределением БАВ по матрице, способом приготовления матрикса, его архитектоникой, способом взаимодействия вещества с носителем – скаффолдом, а также пористостью полимерных конструкций.
Микроскопия – метод анализа необходимый для оценки пористости получаемых полимерных матриксов и пленок, в частности оценки размеров и формы пор, распределения пор по размерам [30, 43, 77, 95]. Наиболее доступным и часто используемым методом является оптическая микроскопия. Сканирующая электронная микроскопия является более информативным, но и более дорогостоящим методом исследования. Она позволяет наблюдать за изменениями морфологии поверхности полимерных структур, размера и формы пор в ходе деградации и гидролиза матрицы [6, 77, 79, 80, 95–97]. При использовании в качестве допантов флуоресцентных зондов для изучения полимерных структур можно использовать флуоресцентную микроскопию (см., например, [98–100]). Она позволяет измерять распределение фотоактивных соединений по полимерной матрице, а также наблюдать за изменениями распределения флуоресцентных зондов по матрице по мере ее деградации.
Спектрофотометрия является наиболее широко используемым методом для установления закономерностей высвобождения молекул БАВ из полимерных структур, в частности, для построения кинетических кривых зависимости количества высвободившегося допанта от времени. В зависимости от химической природы БАВ используют разные оптические диапазоны: УФ-спектроскопию [87, 101, 102] и спектроскопию в УФ-видимой области [103–106]. Процесс высвобождения молекул допанта из полимерной матрицы можно исследовать и с применением флуоресцентных методов [95]. Часто оптические и хроматографические методы применяют совместно. Так в работах [79, 81, 82, 84, 107–110] использовали системы высокоэффективной жидкостной хроматографии (ВЭЖХ) и последующего УФ-детектирования.
Механизмы процессов высвобождения вещества из матрицы можно разделить на две группы: те, что определяют способ высвобождения БАВ (их еще называют “истинными” механизмами) и механизмы, которые позволяют объяснять и предсказывать скорость высвобождения допантов на разных этапах [111]. В зависимости от особенностей взаимодействия полимера, из которого создана матрица, с введенным в нее веществом и растворителем выделяют четыре “истинных” механизма высвобождения допантов [111]:
• осмотическая откачка (природа процесса – конвекция растворителя в материале);
• эрозия (диффузии введенного вещества не происходит, разрушение матрицы лимитирует процесс высвобождения);
• диффузия по полимерному материалу;
• диффузия через поры, заполненные жидкостью.
Механизм осмотической откачки при высвобождении допантов из полилактидных и полилактогликолидных матриц, как правило, реализуется редко [111]. Основным механизмом высвобождения допантов из пористых матриц на основе полиэфиров является диффузия через поры, заполненные жидкостью. Действительно, формы кривых высвобождения молекул допантов из пористых полимерных матриц в различных экспериментах схожи (см., например, рис. 6а) [73, 112–117]. Как правило, скорость высвобождения снижается со временем. В работе [118] представлена кривая высвобождения зонда для пористого матрикса из PDLLA, допированного нитроксильным радикалом TEMPONE, созданного с применением сверхкритических флюидных технологий на основе скCO2. Зонд постепенно высвобождается из полимерной матрицы в течение ~40 дней. Кинетическая кривая высвобождения допанта подчиняется закону Фика (рис. 6б, в). Это и означает, что диффузия через внутренние поры лимитирует высвобождение вещества из матрицы. Было выявлено, что полное высвобождение молекул нитроксильного радикала происходит задолго до момента разрушения полимерной структуры.

Формы кривых высвобождения молекул допантов из полимерных пленок значительно различаются [79, 81, 82, 84, 87, 103–108, 110, 119]. Есть несколько типов кривых: с периодом индукции/без периода индукции, S-образные, с начальным выбросом/без начального выброса вещества (рис. 7) [111, 120]. Предполагают, что и в этом случае реализуется механизм диффузии через поры, но их образование, рост и закрытие на разных этапах играют различную роль [111]. В то же время нельзя исключать и диффузию малых гидрофобных молекул через набухший полимер [121, 122], а также механизм эрозии. Таким образом, мы переходим к описанию механизмов высвобождения, определяющих его скорость на разных этапах.
Рис. 7.
Изображение типичных кинетических кривых высвобождения низкомолекулярных веществ из полимерных матриц в водную среду; шкала времени приведена в условных единицах [120].

Кривая I на рис. 7 иллюстрирует процесс высвобождения низкомолекулярного вещества из однородной среды по закону Фика [123]. Именно так, например, происходит высвобождение малой гидрофобной молекулы соединения bodipy из сополимера PLGA [100], переход которой в поры, заполненные водой, невыгоден. Эта модель также описывает высвобождение допантов из набухших полимерных матриц в случае отсутствия изменений в архитектонике и структуре матрицы на протяжении эксперимента. Так, высвобождение из тонких пленок (<50 мкм) на основе алифатических полиэфиров обычно происходит без периодов индукции в отличие от более толстых образцов (рис. 8).
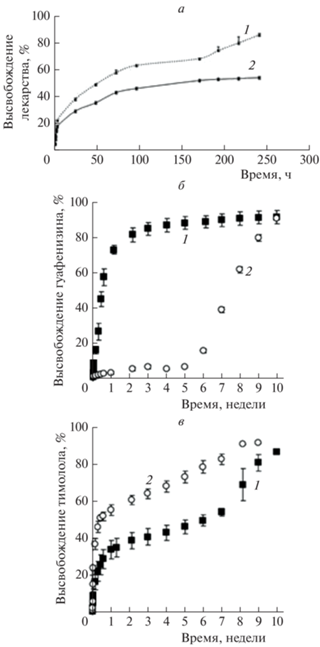
Кривая II (рис. 7) описывает двухэтапный процесс – сначала быстрый “выброс” вещества, затем высвобождение, которое может протекать с постоянной скоростью. Кривая III иллюстрирует трехэтапный процесс высвобождения вещества из матрицы. Первый этап – быстрый “выброс” допанта (иногда эта стадия отсутствует), затем высвобождение введенного вещества существенно замедляется. На третьем этапе высвобождение ускоряется вновь.
Быстрое высвобождение БАВ на начальных этапах процесса высвобождения из алифатических полиэфиров происходит достаточно часто. Оно обычно нежелательно, поскольку сокращает общую продолжительность терапевтического эффекта лекарственного средства, а чрезмерно быстрое высвобождение может вызвать токсическое действие на организм. Часто его объясняют наличием адсорбированнного на поверхности или в порах БАВ, не включенного в полимерную матрицу [124]. Однако многие системы характеризуются равномерным распределением допанта по образцу, что не согласуется с этой моделью.
Многие авторы полагают, что высвобождение на начальном этапе может также протекать через быстро образующиеся в результате сорбции воды приповерхностные поры [77, 125, 126]. Скорость высвобождения можно контролировать, изменяя размер матриц, пористость, концентрацию и природу введенного соединения, а также молекулярную массу полимера.
Рассмотрим влияние этих факторов на примере микрочастиц из PLGA [124]. Показано, что высвобождение допантов из пористой системы происходит намного быстрее, чем из непористого аналога, поскольку пористые микрочастицы имеют большую площадь поверхности и более короткие расстояния для диффузии. Эффект резкого высвобождения на начальном этапе усиливается для больших концентраций введенного вещества, так как в этом случае наблюдается больший начальный градиент концентрации вещества между полимером и средой. При низких концентрациях введенного вещества этот эффект обычно незначителен. Скорость начального выброса контролируется скоростью десорбции лекарственного средства из полимерной матрицы, т.е. растворением вещества с поверхностей частиц в объемной водной фазе. Для гидрофильных лекарств этот эффект проявляется сильнее, чем для гидрофобных из-за их более высокой растворимости в воде. При постоянном размере частиц и концентрации допанта PLGA с более низкой молекулярной массой поглощает значительно большее количество воды, что облегчает диффузию лекарства через полимерную матрицу.
Однако поглощение воды приводит к набуханию и гидролизу полимера, а, следовательно, к снижению его молекулярной массы и увеличению подвижности полимерных цепей, что, в свою очередь, способствует “зарастанию” пор [77, 127]. Этот эффект особенно ярко проявляется в случае более гидрофильного PLGA. Процессы “зарастания” пор зависят от температуры, pH, молекулярной массы полимера и его гидрофобности. “Зарастание” пор для пленок из PLGA наблюдалось как для низких, так и нейтральных значений pH, однако механизм действия был различным. В случае pH = 3 механизм “зарастания” основан на полимер–полимерном взаимодействии, в котором притяжение двух относительно высокогидрофобных областей, разделенных водными взаимодействиями, вызывало высвобождение связанной с поверхностью воды, увеличивая энтропию системы. При pH = 7.4 механизм “зарастания” пор основан на полимер–водном взаимодействии: поры в низкомолекулярном полимере с низкой гидрофобностью закрываются за счет диффузии подвижных полимерных цепей, образуя более набухшую и гомогенную структуру полимера. Этот эффект зависит от температуры: чем выше температура, тем более подвижны полимерные цепи, тем быстрее происходит “зарастание” пор. “Зарастание” протекает интенсивнее у низкомолекулярных и негидрофобных полимеров. В случае сильно гидрофобных полимеров с высокой молекулярной массой “зарастание” пор не происходит. Если исходная пористость матрицы была высокой, то вещество быстро высвободится, в случае непористой структуры основное высвобождение будет происходить на этапе деградации матрицы.
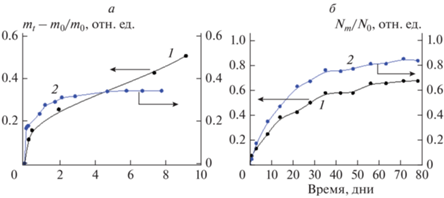
Именно уменьшением количества открытых пор объясняют в работе [77] замедление высвобождения нитроксильных радикалов TEMPOL и ATI из пленок PDLLA толщиной 200 мкм (рис. 9а). При этом проникновение воды в полимерную матрицу не останавливается (рис. 10), что способствует автокаталитическому протеканию гидролиза во внутренних областях пленки, образованию новых, внутренних пор, которые растут и в результате соединяются с поверхностью. Нейтральная среда, окружающая приповерхностный слой пленки, напротив, тормозит гидролиз, что приводит к заметной разнице в температурах стеклования разных зон полимерной матрицы. Действительно, ранее Парк в работе [128] обнаружил, что при выдерживании в воде частицы PLGA характеризуются двумя температурами стеклования, одна из которых (меньшая), со временем уменьшается, а бóльшая – практически не меняется. В работе [77] методом ЭПР установлено, что спиновый зонд выходит из полимерной матрицы в водный раствор в порах, при этом практически на всех этапах имеет место бимодальное распределение зонда по подвижности: быстро вращающиеся частицы в порах и иммобилизованные молекулы в полимерной матрице.
Рис. 9.
а – Дифференциальные кривые высвобождения допантов из пленок PDL02: TEMPOL/200 мкм/76 мм2 (1, черные символы), TEMPOL/200 мкм/55 мм2 (2, красные символы) и ATI/200 мкм/75 мм2 (3, синие символы); б – TEMPOL/50 мкм/14.9 мм2. Линии являются результатом полиномиальной аппроксимации [77].
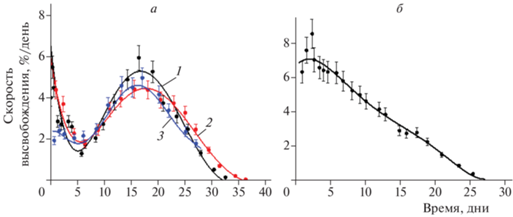
Рис. 10.
Зависимости показателя набухания (1, черные символы) и доли радикалов, локализованных в порах (2, синие символы), от времени для TEMPOL в пленке PDL02 (а) и для TEMPONE в пленке PDL04 (б). Линии не являются результатом математического моделирования, даны для ясности [77].
Смыкание внутренних пор и их выход на поверхность способствует ускорению высвобождения БАВ из эродировавших пленок. В случае аналогичных пленок толщиной 50 мкм кривые высвобождения практически линейны (рис. 9б).
Итак, на первом этапе высвобождение происходит через приповерхностные поры, которые в более толстых пленках затем частично “зарастают”. Скорость высвобождения зонда снижается по мере “зарастания” пор. В это же время в результате локального закисления матрицы внутри нее деградация полимера протекает более интенсивно, чем с поверхности, и зонд высвобождается во внутренние поры. На третьем этапе основной вклад в высвобождение вносит диффузия через образовавшиеся крупные внутренние поры, связанные с поверхностью.
Математическое описание таких процессов является непростой задачей, и некоторые авторы прибегают к феноменологическому описанию кинетических кривых. Так, например, в работах [129, 130] приводится описание кинетики высвобождения допанта, включающее дополнительный параметр – время задержки (tlag), отмечающее начало высвобождения вещества. В работе [131] вариант модификации предполагает учет резкого начального “выброса” вещества. Кроме этого, модель соответствующей закону Фика диффузии, можно модифицировать с учетом релаксационного вклада. Он описывает высвобождение вещества за счет релаксации полимерных цепей, вызванной пластифицирующим действием растворителя [132]. Так как на разных временных интервалах механизмы высвобождения могут быть различными, то разные участки кинетической кривой можно описывать в рамках соответствующих моделей, а затем “сшивать” между собой фрагменты описания [133, 134]. Другой способ – использование коэффициента диффузии как параметра, зависящего от времени [135–139]. Действительно, выдерживание образца в жидкости приводит к его набуханию, меняется свободный объем полимера, упаковка цепей и т.д. В случае полилактидов и полилактогликолидов, кроме этого, нужно учесть автокаталитическое протекание гидролиза, что приводит к изменению морфологии и архитектоники образцов, а также к уменьшению средней молекулярной массы полимера. Помимо этого, можно вводить зависимость коэффициента диффузии не только от времени, но и от пространственных координат [140]. Это позволяет описывать высвобождение допантов из пространственно-неоднородных образцов. Таким образом, можно учесть изменение коэффициента диффузии, вызванное набуханием полимера.
Однако многие из предлагаемых моделей не могут достоверно описать экспериментальные данные во всем временном интервале для матриксов разной геометрии. Кроме того, используемые параметры часто не имеют явного физического смысла, поэтому не дают информации о природе процессов, протекающих в результате гидролиза и набухания полимера, высвобождении допанта из матрицы.
Математическая модель, приведенная в работе [77], лишена многих из этих недостатков. В ней высвобождение спинового зонда из пленок на начальном этапе рассматривали как диффузию, соответствующую закону Фика, без подробного рассмотрения каждого процесса, происходящего с матрицей (диффузия воды в полимер, гидролиз полимерных цепей, диффузия растворимых олигомеров наружу, образование и рост пор, закрытие части пор на поверхности). Чтобы учесть изменения в полимерной матрице в процессе набухания и гидролиза, авторы ввели зависимость коэффициента диффузии от времени и координаты, ${{D}_{{\text{I}}}}\left( {x,t} \right){\text{:}}$
Для описания кинетики высвобождения допанта на втором этапе использовали диффузионную модель, описывающую высвобождение введенного в полимерную матрицу вещества через протяженные поры, связанные с поверхностью и заполненные жидкостью [135]. Молекулы допанта диффундируют через полимерную матрицу с коэффициентом диффузии DS, проникают в поры и диффундируют в жидкость внутри пор с коэффициентом диффузии DL. Эта модель опирается на два основных постулата:
• спиновый зонд имеет две области локализации: полимерная матрица и поры;
• соотношение концентраций зонда в порах и в полимере является постоянным.
Одним из основных параметров модели является функция пористости φ(t), которая представляет собой объемную долю пор, образованных в определенный момент времени и связанных с поверхностью образца (открытых пор). Сначала она была представлена следующим образом:
Данная диффузионная модель хорошо описывает кинетические кривые высвобождения допанта. Однако она использует 11 параметров – слишком большой набор для моделирования экспериментальной зависимости с небольшим количеством особенностей. В связи с этим диффузионная модель была упрощена без ущерба для качества описания экспериментальных данных, с учетом условий решено дифференциальное уравнение и выведено выражение для расчета скорости высвобождения, приведенной к начальному количеству допанта в образце (N0).
Модель может характеризоваться следующими параметрами:
• ${{D}_{0}},~\lambda ,~\mu ,~f$ – эмпирические параметры описания первого этапа высвобождения допантов;
• ${{t}_{{lag}}}$ – определяет начало открытия внутренних пор на поверхности, т.е. начало второго этапа порообразования;
• $A = \unicode{230} {{D}_{L}}{{k}^{{3{\text{/}}2}}}$ – регулирует интенсивность высвобождения допанта на второй стадии порообразования. Данный параметр характеризует сродство допанта к матрице и ее гидролитическую устойчивость (скорость открытия пор на поверхности).
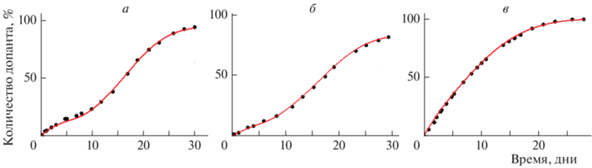
Предложенная модель позволяет моделировать экспериментальные кривые высвобождения спиновых зондов TEMPOL и ATI из пленок PDLLA разной толщины (рис. 11).
Рис. 11.
Экспериментальные значения (точки) и соответствующее численное моделирование (линии) высвобождения TEMPOL (а) и ATI (б) из пленок PDLLA толщиной 200 мкм и TEMPOL (в) из пленки PDLLA толщиной 50 мкм [77].
8. ЗАКЛЮЧЕНИЕ
В данном обзоре отражены современные тенденции в области дизайна матриксов на основе алифатических полиэфиров, а также закономерности их деградации и высвобождения из них биологически активных веществ. Тема эта настолько актуальна, что каждый год появляются не менее ста публикаций, посвященных полимерным матриксам различного состава и строения и отражающих множество разнообразных экспериментальных подходов. Ежегодно предлагаются новые модели для описания сложных процессов, происходящих с полиэфирными материалами in vivo и in vitro, выходят новые обзоры. Данная работа не претендует на полное отображение всех имеющихся достижений в этой области. Помимо описания широко применяемых подходов, основной акцент сделан на использование сверхкритических флюидных технологий для получения полимерных структур, которые могут быть применены в качестве временных протезов или средств контролируемой доставки лекарств и биологически активных добавок. Кроме того, подчеркивается важная роль метода электронного парамагнитного резонанса с применением спинового зонда для характеристики полученных матриксов (пористых скаффолдов и пленок), а также установления закономерностей процессов деградации этих матриксов и высвобождения из них активных компонентов. Активный интерес исследователей к формулировке физических и математических моделей позволяет надеяться, что в ближайшее время появится мощный аппарат, позволяющий предсказывать динамику деградации временных протезов и закономерности высвобождения лекарств и других важных добавок из препаратов пролонгированного действия.
Авторы выражают благодарность П.С. Тимашеву и Н.А. Чумаковой за ценные комментарии и обсуждения в рамках совместной работы. Работа выполнена в рамках государственного задания (регистрационный номер АААА-А21-121011590090-7).
Список литературы
Ramakrishna S., Mayer J., Wintermantel E. et al. // Compos. Sci. Technol. 2001. V. 61. Issue 9. P. 1189.
Vert M. // Biomacromolecules. 2005. V. 6. Issue 2. P. 538.
Ji Y., Ghosh K., Shu X. et al. // Biomaterials. 2006. V. 27. Issue 20. P. 3782.
Piskin E. // J. Biomater. Sci. Polym. Ed. 1995. V. 6. Issue 9. P. 775.
Asghari F., Samiei M., Adibkia K. et al. // Artif. Cells Nanomed. Biotechnol. 2017. V. 45. Issue 2. P. 185.
Iqbal N., Khan A.S., Asif A. et al. // Intern. Mater. Rev. 2019. V. 64. Issue 2. P. 91.
Gunatillake P., Mayadunne R., Adhikari R. // Biotechnol. Annu. Rev. 2006. V. 12. Issue 06. P. 301.
Dhandayuthapani B., Yoshida Y., Maekawa T. et al. // Intern. J. Polym. Sci. 2011. V. 2011. P. 1.
Commandeur S., Van Beusekom H.M.M., Van Der Giessen W.J. // J. Interv. Cardiol. 2006. V. 19. Issue 6. P. 500.
Gentile P., Chiono V., Carmagnola I. et al. // Int. J. Mol. Sci. 2014. V. 15. Issue 3. P. 3640.
Rich J., Korhonen H., Hakala R. et al. // Macromol. Biosci. 2009. V. 9. Issue 7. P. 654.
Rhim J.-W., Mohanty A.K., Singh S.P. et al. // J. Appl. Polym. Sci. 2006. V. 101. Issue 6. P. 3736.
Демина В.А., Седуш Н.Г., Гончаров Е.Н. и др. // Рос. Нанотехнол. 2021. Т. 16. № 1. С. 4.
Lasprilla A.J.R., Martinez G.A.R., Lunelli B.H. et al. // Biotechnol. Adv. 2012. V. 30. Issue 1. P. 321.
Chang L., Liu J., Zhang J. et al. // Polym. Chem. 2013. V. 4. Issue 5. P. 1430.
Vacanti J.P., Langer R. // Lancet. 1999. V. 354. (suppl. 1). P. SI32.
Ma P.X. // Mater. Today. 2004. V. 7. Issue 5. P. 30.
Егорихина М.Н., Мухина П.А., Бронникова И.И. // Комплекс. Пробл. Сердечно-сосудистых заболеваний. 2020. Т. 9. № 1. С. 92.
Chen R.R., Mooney D.J. // Pharm. Res. 2003. V. 20. Issue 8. P. 1103.
Corry D., Moran J. // Biomaterials. 1998. V. 19. Issue 14. P. 1295.
González Corchón M.A., Salvado M., de la Torre B.J. et al. // Ibid. 2006. V. 27. Issue 9. P. 1778.
Freyman T.M., Yannas I. V., Gibson L.J. // Prog. Mater. Sci. 2001. V. 46. P. 273.
Gomes M.E., Reis R.L. // Intern. Mater. Rev. 2004. V. 49. Issue 5. P. 274.
Santos-Rosales V., Iglesias-Mejuto A., Garc C.A. // Polymers (Basel). 2020. V. 12. P. 533.
Vedunova M.V., Timashev P.S., Mishchenko T.A. et al. // Cell Technol. Biol. Med. 2016. V. 161. Issue 4. P. 616.
García-González C.A., Concheiro A., Alvarez-Lorenzo C. // Bioconjugate Chem. 2014. V. 26. Issue 7. P. 1159.
Golubeva E.N., Chumakova N.A., Kuzin S.V. et al. // J. Supercrit. Fluids. 2020. V. 158. P. 104748.
Howdle S.M., Watson M.S., Whitaker M.J. et al. // Chem. Commun. 2001. Issue 1. P. 109.
Antonov E.N., Dunaev A.G., Minaeva S.A. et al. // Pharm. Chem. J. 2018. V. 52. Issue 1. P. 69.
Чумакова Н.А., Голубева Е.Н., Иванова Т.А. и др. // Сверхкритические флюиды: теория и практика. 2018. Т. 13. № 1. С. 86.
Tai H., Mather M.L., Howard D. et al. // Eur. Cells Mater. 2007. V. 14. P. 64.
Song C., Zhang J., Li S. et al. // Chin. J. Chem. Eng. 2021. V. 29. P. 426.
Floren M., Spilimbergo S., Motta A. et al. // J. Biomed. Mater. Res. Part B: Appl. Biomater. 2011. V. 99. Issue 2. P. 338.
Moghadam M.Z., Hassanajili S., Esmaeilzadeh F. et al. // J. Mech. Behav. Biomed. Mater. 2017. V. 69. P. 115.
Santos-Rosales V., Gallo M., Jaeger P. et al. // J. Supercrit. Fluids. 2020. V. 166. P. 105012.
Bhamidipati M., Scurto A.M., Detamore M.S. // Tissue Eng. Part B. Rev. 2013. V. 19. Issue 3. P. 221.
White L.J., Hutter V., Tai H. et al. // Acta Biomater. 2012. V. 8. Issue 1. P. 61.
Reverchon E., Adami R., Cardea S. et al. // J. Supercrit. Fluids. 2009. V. 47. Issue 3. P. 484.
Application of Supercritical Fluidsin Industrial Analysis / Ed. Dean J.R. London: Chapmann&Hall, 1993. P. 1.
Залепугин Д.Ю., Тилькунова Н.А., Чернышова И.В. и др. // Сверхкритические флюиды: теория и практика. 2006. Т. 1. № 1. С. 27.
Зимняков Д.А., Попов В.К., Минаев Н.Б. и др. // Там же. 2020. Т. 15. № 2. С. 27.
Cabezas L.I., Fernández V., Mazarro R. et al. // J. Supercrit. Fluids. 2012. V. 63. P. 155.
Cabezas L.I., Gracia I., García M.T. et al. // Ibid. 2013. V. 80. P. 1.
Залепугин Д.Ю., Тилькунова Н.А., Чернышова И.В. // ССверхкритические флюиды: теория и практика. 2019. Т. 14. № 3. С. 11.
Kroll C., Mäder K., Stößer R. et al. // Eur. J. Pharm. Sci. 1995. V. 3. Issue 1. P. 21.
Mader K. // Biomaterials. 1996. V. 17. Issue 4. P. 457.
Brunner A., Mäder K., Göpferich A. // Pharm. Res. 1999. V. 16. Issue 6. P. 847.
Katzhendler I., Mäder K., Friedman M. // Intern. J. Pharm. 2000. V. 200. Issue 2. P. 161.
Kempe S., Metz H., Mader K. // J. Controlled Release. 2008. V. 130. Issue 3. P. 220.
Kempe S., Metz H., Pereira P.G.C. et al. // Eur. J. Pharm. Biopharm. 2010. V. 74. Issue 1. P. 102.
Kempe S., Metz H., Mäder K. // Eur. J. Pharm. Biopharm. 2010. V. 74. Issue 1. P. 55.
Eisenächer F., Schädlich A., Mäder K. // Intern. J. Pharm. 2011. V. 417. P. 204.
Бучаченко А.Л., Вассерман А.М. Стабильные радикалы. Электронное строение, реакционная способность и применение. М.: Химия, 1973.
Вассерман А.М., Коварский А.Л. Спиновые метки и зонды в физикохимии полимеров. М.: Наука, 1986.
Stoll S., Schweiger A. // J. Magn. Reson. 2006. V. 178. Issue 1. P. 42.
Nitroxides – Theory, Experiment and Applications / Ed. Kokorin A.I. Rijeka, Croatia: InTech, 2012.
Chumakova N.A., Ivanova T.A., Golubeva E.N. et al. // Appl. Magn. Reson. 2018. V. 49. Issue 5. P. 511.
Chumakova N.A., Kuzin S. V., Grechishnikov A.I. // Ibid. 2019. V. 50. № 9. P. 1125.
Nitroxides – Theory, Experiment and Applications / Ed. Kokorin A.I. Rijeka, Croatia: InTech, 2012.
Lurie D., Mader K. // Adv. Drug Deliv. Rev. 2005. V. 57. Issue 8. P. 1171.
Blank A., Freed J.H., Kumar N.P. et al. // J. Controlled Release. 2006. V. 111. Issue 1–2. P. 174.
Mäder K., Bacic G., Domb A. et al. // J. Pharm. Sci. 1997. V. 86. Issue 1. P. 126.
Griffith L.G. // Acta Mater. 2000. V. 48. Issue 1. P. 263.
Merkli A., Tabatabay C., Gurny R. et al. // Prog. Polym. Sci. 1998. V. 23. Issue 3. P. 563.
Morokov E.S., Demina V.A., Sedush N.G. et al. // Acta Biomater. 2020. V. 109. P. 61.
Schliecker G., Schmidt C., Fuchs S. et al. // Biomaterials. 2003. V. 24. Issue 21. P. 3835.
Gorrasi G., Pantani R. // Adv. Polym. Sci. 2018. V. 279. P. 119.
Vert M., Schwarch G., Coudane J. // J. Macromol. Sci. Part A. 1995. V. 32. Issue 4. P. 787.
Aso Y., Yoshioka S., Li Wan Po A. et al. // J. Controlled Release. 1994. V. 31. Issue 1. P. 33.
Heya T., Okada H., Ogawa Y. et al. // J. Pharm. Sci. 1994. V. 83. Issue 5. P. 636.
Saha S.K., Tsuji H. // Polym. Degrad. Stab. 2006. V. 91. Issue 8. P. 1665.
Saha S.K., Tsuji H. // Macromol. Mater. Eng. 2006. V. 291. Issue 4. P. 357.
Cabezas L.I., Gracia I., de Lucas A. et al. // Ind. Eng. Chem. Res. 2014. V. 53. Issue 40. P. 15374.
Siepmann J. // Adv. Drug Delivery. Rev. 2001. V. 48. P. 229.
Holland S.J., Jolly A.M., Yasin M. et al. // Biomaterials. 1987. V. 8. Issue 4. P. 289.
Mäder K., Bittner B., Li Y. et al. // Pharm. Res. 1998. V. 15. Issue 5. P. 787.
Chumakova N.A., Golubeva E.N., Kuzin S.V., Ivanova T.A. et al. // Polymers (Basel). 2020. V. 12. Issue 12. P. 1.
Proikakis C.S., Mamouzelos N.J., Tarantili P.A. et al. // Polym. Degrad. Stab. 2006. V. 91. Issue 3. P. 614.
Frank A., Rath S.K., Venkatraman S.S. // J. Controlled Release. 2005. V. 102. Issue 2. P. 333.
Wan Y., Fang Y., Wu H. et al. // J. Biomed. Mater. Res. Part A. 2007. V. 80. Issue 4. P. 776.
Tarvainen T., Karjalainen T., Malin M. et al. // J. Controlled Release. 2002. V. 81. Issue 3. P. 251.
Tarvainen T., Malin M., Barragan I. et al. // Intern. J. Pharm. 2006. V. 310. P. 162.
Witt C., Mäder K., Kissel T. // Biomaterials. 2000. V. 21. Issue 9. P. 931.
Burke J., Donno R., D’Arcy R. et al. // Biomacromolecules. 2017. V. 18. Issue 3. P. 728.
Tsuji H., Ikada Y. // J. Polym. Sci., Part A: Polym. Chem. 1998. V. 36. Issue 1. P. 59.
Tsuji H. // Polymer (Guildf). 2002. V. 43. Issue 6. P. 1789.
Klose D., Siepmann F., Elkharraz K. et al. // Int. J. Pharm. 2008. V. 354. P. 95.
Grizzi I., Garreau H., Li S. et al. // Biomaterials. 1995. V. 16. Issue 4. P. 305.
Tsuji H., Ikada Y. // Polym. Degrad. Stab. 2000. V. 67. Issue 1. P. 179.
De Jong S.J., Arias E.R., Rijkers D.T.S. et al. // Polymer (Guildf). 2001. V. 42. Issue 7. P. 2795.
Poly(factic acid): Synthesis, Structures, Properties, Processings and Applications / Eds. Auras R., Lim L., Selke S., Tsnji H. Hoboken, NJ, USA: John Wiley & Sons, Inc. 2010. Part IV. Ch. 21.
Fischer E.W., Sterzel H.J., Wegner G. // Kolloid-Zeitschrift und Zeitschrift für Polym. 1973. V. 251. № 11. P. 980.
Maulding H.V., Tice T.R., Cowsar D.R. et al. // J. Controlled Release. 1986. V. 3. P. 103.
Bobko A.A., Kirilyuk I.A., Grigor’ev I.A. et al. // Free Radical. Biol. Med. 2007. V. 42. P. 404.
Xu Q., Chin S.E., Wang C.-H. et al. // Biomaterials. 2013. V. 34. № 15. P. 3902.
Strobel C., Bormann N., Kadow-Romacker A. et al. // J. Controlled Release. 2011. V. 156. Issue 1. P. 37.
Zhang E., Zhu C., Yang J. et al. // Mater. Sci. Eng., C. 2016. V. 58. P. 278.
Determan A.S., Trewyn B.G., Lin V.S.Y. et al. // J. Controlled Release. 2004. V. 100. Issue 1. P. 97.
Kim B.S., Oh J.M., Kim K.S. et al. // Biomaterials. 2009. V. 30. Issue 5. P. 902.
Kang J.C., Schwendeman S.P. // Macromolecules. 2003. V. 36. Issue 4. P. 1324.
Brannon-Peppas L., Peppas N.A. // J. Controlled Release. 1989. V. 8. Issue 3. P. 267.
Fayzullin A., Churbanov S., Ignatieva N. et al. // Biomedicines. 2021. V. 9. Issue 8. P. 853.
Jonnalagadda S., Robinson D.H. // J. Appl. Polym. Sci. 2004. V. 93. Issue 5. P. 2025.
Wang Y., Challa P., Epstein D.L. et al. // Biomaterials. 2004. V. 25. Issue 18. P. 4279.
Wu C., Ramaswamy Y., Zhu Y. et al. // Ibid. 2009. V. 30. Issue 12. P. 2199.
Mashak A., Mobedi H., Mahdavi H. // Pharm. Sci. (Iran) 2015. V. 21. Issue 2. P. 77.
Dong Y., Zhang Z., Feng S.-S. // Intern. J. Pharm. 2008. V. 350. P. 166.
Owen G.R., Jackson J.K., Chehroudi B. et al. // J. Biomed. Mater. Res. Part A. 2010. V. 95. Issue 3. P. 857.
Lee K., Silva E.A., Mooney D.J. // J. R. Soc. Interface. 2011. V. 8. Issue 55. P. 153.
Ma D., McHugh A.J. // Intern. J. Pharm. 2010. V. 388. P. 1.
Fredenberg S., Wahlgren M., Reslow M. et al. // Ibid. 2011. V. 415. P. 34.
Cardea S., Baldino L., Scognamiglio M. et al. // J. Mater. Sci. – Mater. Med. 2014. V. 25. Issue 4. P. 989.
Santo V.E., Duarte A.R.C., Popa E.G. et al. // J. Controlled Release. 2012. V. 162. Issue 1. P. 19.
Nof M., Shea L.D. // J. Biomed. Mater. Res. 2002. V. 59. Issue 2. P. 349.
Richardson T.P., Peters M.C., Ennett A.B. et al. // Nat. Biotechnol. 2001. V. 19. Issue 11. P. 1029.
Duarte A.R.C., Santo V.E., Alves A. et al. // J. Supercrit. Fluids. 2013. V. 79. P. 177.
Velasco D., Benito L., Fernández-Gutiérrez M. et al. // Ibid. 2010. V. 54. Issue 3. P. 335.
Иванова Т.А., Чумакова Н.А., Голубева Е.Н. и др. // Сверхкритические флюиды: теория и практика. 2019. Т. 14. № 1. С. 67.
Shah S.S., Cha Y., Pitt C.G. // J. Controlled Release. 1992. V. 18. Issue 3. P. 261.
Xu Y., Kim C.S., Saylor D.M. et al. // J. Biomed. Mater. Res. Part B. 2017. V. 105. Issue 6. P. 1692.
Raman C., Berkland C., Kim K. et al. // J. Controlled Release. 2005. V. 103. Issue 1. P. 149.
Wischke C., Schwendeman S.P. // Intern. J. Pharm. 2008. V. 364. Issue 2. P. 298.
Siepmann J., Siepmann F. // J. Controlled Release. 2012. V. 161. Issue 2. P. 351.
Yoo J., Won Y. // ACS Biomater. Sci. Eng. 2020. V. 6. Issue 11. P. 6053.
Mochizuki A., Niikawa T., Omura I. et al. // J. Appl. Polym. Sci. 2008. V. 108. P. 3353.
Webber W.L., Lago F., Thanos C. et al. // J. Biomed. Mater. Res. 1998. V. 41. P. 18.
Fredenberg S., Wahlgren M., Reslow M. et al. // J. Controlled Release. 2011. V. 150. Issue 2. P. 142.
Park T.G. // Biomaterials. 1995. V. 16. Issue 15. P. 1123.
Ford L.J., Mitchell K., Rowe P. et al. // Intern. J. Pharm. 1991. V. 71. Issue 1–2. P. 95.
Kao C., Chen S., Sheu M. // J. Controlled Release. 1997. V. 44. Issue 2–3. P. 263.
Lindner W.D., Lippold B.C. // Pharm. Res. 1995. V. 12. P. 1781.
Peppas N.A., Sahlin J.J. // Intern. J. Pharm. 1989. V. 57. Issue 2. P. 169.
Lao L.L., Venkatraman S.S., Peppas N.A. // Eur. J. Pharm. Biopharm. 2008. V. 70. Issue 3. P. 796.
Batycky P.R., Hanes J., Langer R. et al. // J. Pharm. Sci. 1997. V. 86. Issue 12. P. 1464.
Zhu X., Braatz R.D. // J. Biomed. Mater. Res. Part A. 2015. V. 103. Issue 7. P. 2269.
Charlier A., Leclerc B., Couarraze G. // Intern. J. Pharm. 2000. V. 200. Issue 1. P. 115.
Faisant N., Akiki J., Siepmann F. et al. // Ibid. 2006. V. 314. Issue 2. P. 189.
Casalini T., Rossi F., Lazzari S. et al. // Mol. Pharm. 2014. V. 11. Issue 11. P. 4036.
Wada R., Hyon S., Ikada Y. // J. Controlled Release. 1995. V. 37. Issue 1–2. P. 151.
Joshi A., Himmelstein K.J. // Ibid. 1991. V. 15. Issue 2. P. 95.
Baino F. // Acta Biomater. 2011. V. 7. Issue 9. P. 3248.
You Y., Min B.-M., Lee S.J. et al. // J. Appl. Polym. Sci. 2005. V. 95. Issue 2. P. 193.
You Y., Lee S.W., Youk J.H. et al. // Polym. Degrad. Stab. 2005. V. 90. Issue 3. P. 441.
Bergsma J. et al. // Biomaterials. 1995. V. 16. Issue 1. P. 25.
Agrawal C.M., Ray R.B. // J. Biomed. Mater. Res. 2001. V. 55. Issue 2. P. 141.
Sarazin P., Roy X., Favis B.D. // Biomaterials. 2004. V. 25. Issue 28. P. 5965.
Vert M., Li S., Garreau H. et al. // Angew. Makromol. Chem. 1997. V. 247. Issue 1. P. 239.
Hussain Y.A., Grant C.S. // J. Supercrit. Fluids. 2012. V. 71. P. 127.
Ramchandani M., Robinson D. // J. Controlled Release. 1998. V. 54. Issue 2. P. 167.
Duarte A.R.C., Mano J.F., Reis R.L. // Acta Biomater. 2009. V. 5. Issue 6. P. 2054.
Yoganathan R., Mammucari R., Foster N.R. // J. Phys. Conf. Ser. 2010. V. 215. P. 012087.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика