

Химическая физика, 2022, T. 41, № 6, стр. 55-64
Перенос зарядов в биомиметических моделях органических нейроморфных материалов
Л. А. Савинцева 1, *, А. А. Авдошин 1, С. К. Игнатов 1
1 Нижегородский государственный университет им. Н.И. Лобачевского
Нижний Новгород, Россия
* E-mail: savintsevaliana@yandex.ru
Поступила в редакцию 06.12.2021
После доработки 14.01.2022
Принята к публикации 20.01.2022
- EDN: VWYMUK
- DOI: 10.31857/S0207401X22060103
Аннотация
Проведено моделирование процесса переноса заряда в материалах на основе органических полупроводников и биоорганических молекул: дицианвинилолиготиофенов, производных порфинов и хинонов, которые рассматриваются как основа для создания молекулярных искусственных нейронных сетей (ИНС). Квантовохимическими методами оценены энергетические и электронные характеристики этих молекул, определены их параметры в силовом поле OPLS-AA. Динамика заряда моделировалась при помощи кинетического метода Монте-Карло в системах из 1200 молекул, уравновешенных методом молекулярной динамики с оценкой констант туннелирования по теории Маркуса. На основе сравнения подвижности заряда в моделируемых системах обсуждаются возможные пути модификации исследованных молекул для создания ИНС.
1. ВВЕДЕНИЕ
Нервная система живых организмов функционирует путем передачи электрохимических сигналов между нейронами, образующими разветвленную сетевую структуру. Возникает вопрос о возможности создания искусственного материала, в котором зарядовый транспорт между молекулами воспроизводил бы передачу сигнала в нейронной сети, а, следовательно, и поведение сети нейронов на молекулярном уровне. Такой материал, если его рассматривать как вычислительную систему (искусственную нейронную сеть (ИНС)), позволил бы добиться высокой плотности вычислительных элементов, их трехмерной организации и высокой вычислительной эффективности.
Для функционирования в качестве ИНС молекулы материала должны обладать следующими свойствами: 1) высокой способностью к межмолекулярной передаче заряда; 2) функцией “суммирования” сигнала от разных источников; 3) возможностью изменения межмолекулярных расстояний в случае реакции на повторяющиеся сигналы. Целью настоящей работы является поиск материалов, способных к проявлению таких “нейроморфных” свойств. В данной работе мы фокусируем внимание на поиске разветвленных молекул, обладающих первым свойством и частично – вторым, т.е. высокой способностью к внутри- и межмолекулярной передаче носителей заряда, а также структурой в виде “ядро с ответвлениями”. Такая структура потенциально способна осуществлять накопление заряда на центральном ядре, что является основой для осуществления логических операций (например, дальнейшей передачи сигнала в зависимости от заряда “ядра”).
Поскольку в природе известны молекулы, участвующие в процессах электронного переноса, разумно использовать их в качестве потенциальных кандидатов для поиска молекул нейроморфного материала. Подход, основанный на биомиметических принципах, часто используется в современных нанотехнологиях (см., например, [1]). В данной работе мы рассматриваем несколько представителей таких биомиметических систем, являющихся производными хинонов и порфиринов, в которых изучена зарядовая проводимость [2–5], а также фталоцианин, широко использующийся в качестве фотокатализатора и полупроводникового материала [6], фитол и азотистые основания, входящие в состав ДНК (РНК) живых организмов. С целью сопоставления рассчитываемых электрофизических свойств с экспериментальными данными мы также рассматриваем в качестве реперных молекул известные органические полупроводники, относящиеся к классу дициановинилолиготиофенов DCVnT [7–9], полициклические ароматические углеводороды, нормальные π-сопряженные углеводороды. Для рассматриваемых молекул мы проводим расчеты молекулярной структуры, электронных параметров, влияющих на электронный перенос (потенциалов ионизации и энергий фронтальных орбиталей), а в случае двух систем – характеристик зарядовой проводимости.
2. ТЕОРИЯ И МЕТОДЫ
2.1. Оценка кинетики элементарного акта туннелирования
Процесс переноса заряда в органических полупроводниках осуществляется последовательными квантовыми прыжками (туннелированием) между центрами локализации электрона – донорами и акцепторами [10]. В качестве центров локализации обычно рассматриваются локализованные молекулярные орбитали (ЛМО) фрагментов, выбранных, например, в соответствии с процедурой локализации NBO. В зависимости от расположения центров локализации возможны внутримолекулярный и межмолекулярный переносы заряда. Вероятность процесса перехода электрона с ЛМО одного фрагмента на ЛМО другого фрагмента высока: во-первых, если они пространственно близки, и, во-вторых, если близки энергии этих ЛМО. Выравнивание энергий ЛМО двух фрагментов может произойти за счет теплового колебательного движения ядер молекулы. Этот же процесс обеспечивает необратимость переноса электрона, поскольку вследствие ядерной релаксации сразу же после туннелирования энергии двух ЛМО станут неэквивалентными и обратный процесс будет запрещен. В результате этого процесс переноса заряда тесно связан с колебаниями молекул, их конформациями, а также с наличием пространственных контактов между центрами локализации. Началу процесса соответствует электронное возбуждение за счет, например, внешнего электромагнитного воздействия. В ходе процесса возможны два альтернативных механизма: электронный и дырочный перенос [11].
Скорость процесса переноса заряда описывается различными теориями [12, 13], среди которых компромисс между теоретической строгостью и вычислительной сложностью достигается в случае высокотемпературного предела классической теории – теории Маркуса [14]. В этой теории константа скорости переноса дается следующим аналитическим выражением:
Интеграл переноса является мерой силы электронной связи в начальном и конечном состояниях в комплексе с переносом заряда. Обычно его оценивают с использованием эффективного одночастичного гамильтониана:
Изменение ядерных степеней свободы вследствие перемещения заряда от донора к акцептору учитывается в энергии реорганизации. Ее физический смысл состоит в том, что она представляет собой энергию, затрачиваемую на “подстройку” молекулярного окружения донора и акцептора для осуществления туннелирования между ними, и содержит два вклада: внутримолекулярный, который обусловлен перестройкой ядерных координат двух молекул, образующих комплекс с переносом заряда, и внешнесферный, который обусловлен релаксацией ядерных координат среды [16].
В общем случае поверхности потенциальной энергии (ППЭ) донора и акцептора не идентичны, поэтому внутримолекулярные энергии реорганизации различаются. Для случая разрядки (т.е. потери электрона) молекулы $i$ и зарядки молекулы $j$ можно записать $\lambda _{{ij}}^{{int}} = \lambda _{i}^{{cn}} + \lambda _{j}^{{nc}}$ = $U_{i}^{{nC}} - U_{i}^{{nN}} + U_{j}^{{cN}} - U_{j}^{{cC}}.$ Здесь $U_{i}^{{nC}}$ – это внутренняя энергия нейтральной молекулы $i$ в геометрической структуре ее заряженного состояния (индекс “n” обозначает основное состояние, а индекс “С” – структуру заряженной молекулы). Аналогично, $U_{j}^{{cN}}$ – это энергия заряженной молекулы $j$ в геометрии нейтрального состояния. Поверхности потенциальной энергии донора и акцептора неидентичны для химически разных соединений или конформеров одной молекулы, поэтому в общем случае $\lambda _{i}^{{cn}} \ne \lambda _{j}^{{cn}}$ и $\lambda _{i}^{{nc}} \ne \lambda _{j}^{{nc}}.$
2.2. Квантовохимическая оценка энергии ЛМО
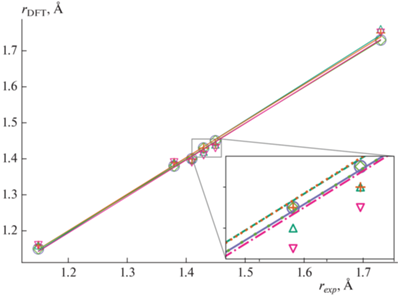
Для правильной и быстрой оценки энергии центров локализации важно корректно рассчитывать энергии электрона и дырки (т.е. катионного и анионного состояний молекул). Такая оценка для молекул в составе материала возможна по теореме Купманса на основе величины энергии МО. Однако не все квантовохимические методы обеспечивают хорошее согласие между энергиями ионизации, рассчитанными по теореме Купманса, и энергиями ионных состояний. Для выбора метода была проведена серия расчетов структуры и потенциалов ионизации молекул с применением четырех функционалов DFT и одного композитного метода электронной структуры – r2-SCAN-3c. Рассмотренные уровни теории представлены на рис. 1, где проведено сопоставление результатов расчета геометрической структуры и экспериментальных данных для ряда реперных молекул. Видно, что все методы достаточно хорошо воспроизводят экспериментальные значения длин связей для молекул DCVnT.
Рис. 1.
Сопоставление рассчитанных (rDFT) и экспериментальных (rexp) длин связей для молекул дицианвинилолиготиофенов DCVnT следующими методами: △ – B3LYP/6-31G(d, p), RMSD = 0.015; + – B3LYP/6-311+G(d, p), RMSD = = 0.012; ⚪ – M062X/cc-pVTZ, RMSD = 0.004, ◇ – wB97XD/cc-pVTZ, RMSD = 0.004; ▽ – r2-SCAN, RMSD = 0.017.
На рис. 2 приведено сопоставление расчетных и экспериментальных данных для потенциалов ионизации (IP), сродства к электрону (AE) и ширины запрещенной зоны (Egap), которые определяют электронные свойства молекул и входят в формулу для расчета энергии реорганизации. Известно, что методы DFT могут давать большой разброс при расчете энергетических параметров. В работе [17] показано, что meta-GGA-функционалы M06-2X и M11 хорошо воспроизводят значения IP для широкого диапазона органических молекул, поэтому была проведена серия расчетов с использованием этих функционалов, а также современного метода r2-SCAN-3c, который должен обеспечивать лучшее воспроизведение электронных свойств.
Рис. 2.
Сравнение результатов расчета: а – потенциалов ионизации и сродства к электрону с экспериментальными данными для тестового набора данных (○ – M062X/cc-pVTZ, RMSD = 0.621; △ – M11/cc-pVTZ, RMSD = 0.627; ☆ – r2-SCAN, RMSD = = 0.485); б – энергий ε граничных орбиталей (высшей занятой и низшей свободной) с рассчитанными величинами IP и EA для тестового набора данных (⚪ – M062X/cc-pVTZ, RMSD = 1.218; △ – M11/cc-pVTZ, RMSD = 0.222; ☆ – r2-SCAN, RMSD = = 2.950); в – значений IP и EA, полученных методом M062X/cc-pVTZ, с экспериментальными величинами для полного набора данных при RMSD = 0.376.
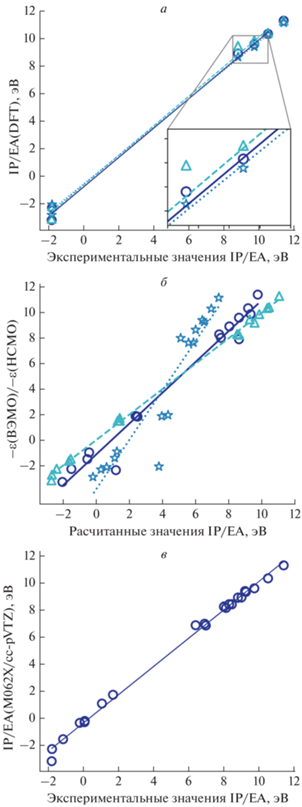
Анализ результатов расчета показывает, что метод r2-SCAN-3c хорошо воспроизводит значения IP и AE, хотя и уступает другим методам в точности воспроизведения геометрической структуры. Однако значения энергий граничных орбиталей плохо коррелируют со значениями IP и AE, что затрудняет электронный анализ исследуемых структур. Таким образом, для дальнейших расчетов был выбран метод M062X/cc-pVTZ как метод, обеспечивающий компромисс между точностью и вычислительными затратами.
2.3. Молекулярно-динамическое моделирование структуры органического материала
Молекулярно-динамическое моделирование (МД) осуществлялось с использованием силового поля OPLS-AA, изначально разработанного для белковых молекул. Однако ряд изучаемых молекулярных фрагментов не имеют параметров в этом силовом поле, что требует его дополнительной параметризации. Для параметризации силового поля молекулы были разбиты на жесткие фрагменты, которые в терминологии силовых полей называются остатками и составляют так называемые сопряженные сегменты. Один сопряженный сегмент содержит несколько жестких фрагментов.
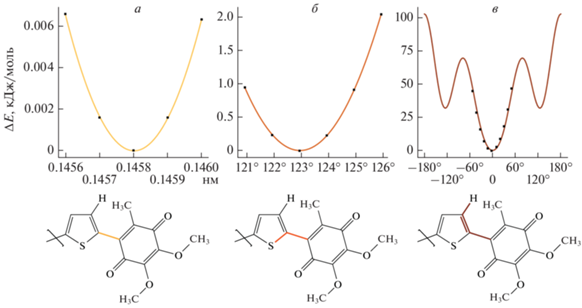
Для выбранных фрагментов при помощи открытой онлайн-программы TPPmktop [18] была построена топология и определены константы невалентных (несвязанных) взаимодействий, а также тех валентных взаимодействий, которые аналогичны существующим в силовом поле. Для оставшихся валентных взаимодействий силовые константы были определены путем сканирования ППЭ изучаемых молекул при изменении длины связей, валентных и торсионных углов. Примеры проведенных вычислений для одной из изучаемых молекул приведены на рис. 3.
Рис. 3.
Вид сканируемых ППЭ (вверху) молекулярного фрагмента с указанием сканируемых геометрических параметров (внизу): а – потенциал изменения длины связи в нм ($161379{{x}^{2}} - 47095x + 347,$ константа растяжения $k = 2 \cdot 161379 = 3.2 \cdot {{10}^{5}}$) кДж/моль); б – гармонический потенциал изменения валентного угла в град ($0.2298{{x}^{2}} - 57x + 3474,$ константа $k = 2 \cdot {{\left( {{{180} \mathord{\left/ {\vphantom {{180} \pi }} \right. \kern-0em} \pi }} \right)}^{2}} \cdot 0.2298 = 1510$ кДж/моль; в – периодическая функция изменения торсионного угла в град (Ryckaert-Bellemans function) при ${{C}_{0}} = 64.6,$ ${{C}_{1}} = - 48.8,$ ${{C}_{2}} = - 100.0,$ ${{C}_{3}} = 100.0,$ ${{C}_{4}} = 86.9,$ ${{C}_{5}} = 0.0$ (в кДж/моль).
Аморфный органический материал моделировался молекулами исследуемых олигомеров, случайным образом упакованных в элементарной ячейке. Элементарная ячейка представляла собой кубический бокс с наложенными периодическими граничными условиями, содержащий 1000–1200 молекул с геометрическими параметрами, оптимизированными методом DFT. Система уравновешивалась в состоянии NVT-ансамбля в течение 0.5 нс с шагом по времени 0.001 пс (термостат типа V-rescale), а затем в состоянии NPT-ансамбля в течение 10 нс (баростат Парринелло–Рахмана, T = 300 K). В расчетах использовалось силовое поле OPLS-AA, парамтеризованное в настоящей работе.
2.4. Моделирование переноса заряда
Динамика носителей заряда моделировалась путем оценки скорости межмолекулярных перескоков носителей заряда и решения основного уравнения для диффузии носителей заряда в приложенном электрическом поле с использованием инетического метода Монте-Карло, реализованного в программном пакете VOTCA-CTP [19, 20].
Внутримолекулярная энергия реорганизации была рассчитана квантовохимическим методом на основе величины энергии ионизированных состояний молекул. Например, при движении дырки (hole (h)) энергия реорганизации выражается как
Предполагая, что перенос заряда намного медленнее, чем электронная поляризация, но намного быстрее, чем перестройка ядер среды (вне комплекса), внешнесферная энергия реорганизации $\lambda _{{ij}}^{{out}}$ может быть рассчитана по величинам полей электрического смещения, создаваемых комплексом с переносом заряда [21]:
Для расчета интегралов переноса был использован метод ZINDO [22], реализованный в программе VOTCA-CTP. Метод ZINDO позволяет избежать самосогласованных расчетов для каждого отдельного мономера и димера, что дает возможность построить матричные элементы гамильтониана для димера из взвешенного перекрытия молекулярных орбиталей двух мономеров. Вместе с введением жестких сегментов требуется только один самосогласованный расчет для одного изолированного сопряженного сегмента. Все соответствующие молекулярные перекрытия могут быть построены из полученных молекулярных орбиталей.
На основе анализа структуры материала, полученного в ходе МД-моделирования, были определены список сопряженных сегментов (мест перескоков) и скорости передачи заряда между ними. Динамика носителей заряда моделировалась путем оценки скорости их межмолекулярных перескоков и решения основного уравнения для диффузии носителей заряда в приложенном электрическом поле с использованием кинетического метода Монте-Карло. Основное уравнение, описывающее эволюцию системы во времени, записывается как
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 4 приведены значения энергий граничных орбиталей для всех исследуемых молекул, определенные методом M062X/cc-pVTZ: на рис. 4а – значения для “ядер” конструируемых молекул, на рис. 4б – для молекул малого размера, рассматриваемых в качестве коротких боковых цепей; на рис. 4в – для олигомеров, рассматриваемых в качестве длинных боковых цепей. Видно, что большинство исследуемых молекул являются диэлектриками. Такие молекулы, как антрацен, порфин, хлорин, тетрааллилпорфин, фталоцианин, дицианвинилдитиофен и сопряженные углеводороды, являются полупроводниками с шириной запрещенной зоны от 3.4 (фталоцианин) до 5.1 эВ (дицианвинилдитиофен). Исходя из значений ширины запрещенной зоны и энергий граничных орбиталей, для моделирования процесса переноса заряда были выбраны производные убихинона (U10) и порфина (P). Дополнительно были рассмотрены аморфные материалы на основе дициановинилдитиофена DCV2T, поскольку для этого органического полупроводника накоплено большое количество экспериментальных данных, что позволяет рассматривать его в качестве реперной системы. Молекулы, для которых проводилось МД-моделирование, представлены на рис. 5. Количество молекул в моделируемых системах составляло 1000 в системе тетрааллилпорфина (TAP) и 1200 в системах дицианвинилдитиофена (DCV2T) и соединения убихинона с дицианвинилдитиофеном (U10–CV2T).
Рис. 4.
Энергии граничных орбиталей исследуемых молекул: а – 1 – тиофен, 2 – бензол, 3 – нафталин, 4 – антрацен, 5 – тетрацен, 6 – аденин, 7 – гуанин, 8 – цитозин, 9 – тимин, 10 – урацил, 11 – порфин, 12 – хлорин, 13 – тетрафенилпорфин (TAP), 14 – фталоцианин, 15 – убихинон (U10), 16 – пластохинон, 17 – дигидропиридин; б – 1 – ацетилен, 2 – этилен, 3 – пропилен; в – 1 – дицианвинилдитиофен (CV2T), 2 – фитол, 3 – 2-метилдекан, 4 – 2,6-диметилдека-1,3,5,7,9-пентаен, 5 – 2,6,10-триметилдодека-1,3,5,7,9,11-гексаен.

Рис. 5.
Структуры исследуемых молекул (сверху) и результаты моделирования (снизу): а и б – МД и переноса заряда; в – фрагмент моделируемого бокса с иллюстрацией π-стэкинга между молекулами TAP.

Рассчитанные внутримолекулярные энергии реорганизации представлены в табл. 1. Очевидно, что чем меньше энергия реорганизации, тем легче должен протекать процесс переноса заряда в комплексе. Наименьшие значения имеют системы DCV2T и P–CV2T (молекула порфина, к одному из колец которой присоединен дицианвинилдитиофен). Из данных табл. 1 также следует, что энергии реорганизации выше в случае переноса электрона, чем в случае переноса дырки, и это имеет место для всех систем, кроме P–CV2T, где они приблизительно равны. Таким образом, перенос дырок должен быть более выгоден, чем перенос электрона.
Таблица 1.
Рассчитанные внутримолекулярные энергии реорганизации
| Молекула | Носитель заряда | λ1, эВ | λ2, эВ |
|---|---|---|---|
| DCV2T | дырка | 0.0834 | 0.0845 |
| электрон | 0.1520 | 0.1516 | |
| DCV4T | дырка | 0.0985 | 0.1124 |
| электрон | 0.1320 | 0.1364 | |
| U10–CV2T | дырка | 0.1506 | 0.1872 |
| электрон | 0.1506 | 0.2344 | |
| TAP | дырка | 0.1020 | 0.0971 |
| электрон | 0.1104 | 0.1207 | |
| P–CV2T | дырка | 0.0691 | 0.0663 |
| электрон | 0.0634 | 0.0684 |
Начальные условия и результаты МД-моделирования приведены в табл. 2. В случае DCV2T рассчитанная плотность аморфного материала составляет 1257 кг/м3, что близко к значению плотности кристаллического DCV4T, равной 1334 кг/м3 [23]. Этот факт свидетельствует о правильном воспроизведении силовым полем типичных межмолекулярных расстояний в аморфном полимерном материале.
Таблица 2.
Рассчитанные структурные и физические параметры аморфных материалов, изученных методом МД
| Молекула | Начальные условия | Конечные условия | ||
|---|---|---|---|---|
| размер ячейки, нм | плотность, кг/м3 | размер ячейки, нм | плотность, кг/м3 | |
| DCV2T | 9.6 × 10.0 × 9.8 | 680.15 | 7.8 × 8.2 × 8.0 | 1257.2 |
| U10–CV2T | 12.8 × 10.0 × 9.4 | 668.9 | 10.4 × 8.1 × 7.6 | 1262.6 |
| TAP | 33.4 × 33.3 × 23.0 | 26.9 | 19.4 × 19.4 × 13.4 | 136.5 |
В случае материала на основе порфина оказалось, что органические молекулы образуют “стопки” с большим расстоянием между ними (рис. 5в) в результате π-стекинга [24], что приводит к очень низкой плотности материала. Плотность внутри отдельной стопки, определенная интегрированием по объему с шагом интегрирования 0.3 × 0.3 × 0.3 нм3 (это значение получено дифференцированием зависимости относительного объема ячейки от величины шага), составляет 1064.5 кг/м3, плотность порфина без аллильных заместителей – 1350 кг/м3. По-видимому, при описании молекул порфина силовым полем OPLS значение энергии π-стекинга сильно завышается, а латеральные взаимодействия недооцениваются, что не позволяет адекватно описывать аморфные материалы на основе этих молекул без добавления растворителя или объемных заместителей.
В исследуемых системах после МД-симуляции осуществлялся поиск межмолекулярных контактов с расстоянием между центрами масс молекул не более чем 0.8 нм, что соответствует примерно 12-ти соседним молекулам в первой координационной сфере [20]. Эти пары рассматривались как перспективные контакты для межмолекулярного переноса заряда, для которых вычислялись интегралы и скорости переноса. Типичные значения интегралов переноса для органических полупроводников составляют 10–200 эВ, причем значения в 100 эВ и выше не характерны для большинства органических соединений [25]. В случае переноса электрона в системе DCV2T всего 4.0% пар имеют $J > 10$ эВ, а в случае переноса дырки – 10.2% пар. Для системы U10-CV2T это 3.9 и 5.0% соответственно (рис. 6).
Рис. 6.
Распределение интегралов переноса для дырки (а) и электрона (б) в моделируемых системах (помечены значения ${{J}_{{ij}}} > 10$ эВ).
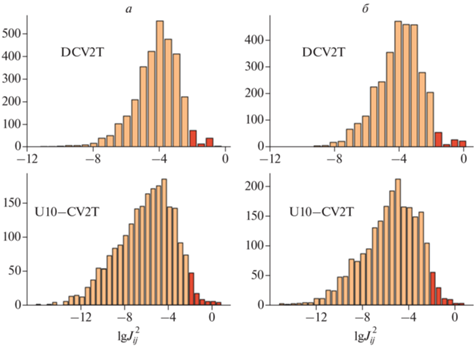
Большие значения интеграла переноса способствуют делокализации заряда и, следовательно, увеличению его мобильности. Большая доля высоких интегралов в случае переноса дырки подтверждает вывод о том, что перенос электрона в обеих системах должен быть более медленным, чем перенос дырки. Этот вывод согласуется с экспериментальными работами [7, 20].
Моделирование зарядового транспорта длительностью ${{10}^{{ - 3}}}$ с для дырки и 1 с для электрона осуществлялось с учетом действия однородного электростатического поля напряженностью ${{10}^{6}}$ В/м, приложенного в направлении оси z. Кинетические характеристики метода Монте-Карло усреднялись по 50-ти траекториям движения заряда. Рассчитанные подвижности носителей заряда приведены в табл. 3.
Таблица 3.
Подвижности зарядов (в см2/В · с), рассчитанные методом КМК
| Носитель заряда | DCV2T | U10–CV2T | |
|---|---|---|---|
| расчет из данной работе | расчет из работы [5] | ||
| Дырка | 2.2 · 10–8 | 1.1 · 10–9 | 7.1 · 10–10 |
| Электрон | 3.7 · 10–10 | – | 9.9 · 10–11 |
Как следует из табл. 3, результаты расчета в случае DCV2T свидетельствуют, что в материале преобладает дырочная проводимость, т.е. подвижность электронов почти на два порядка ниже подвижности дырок. Этот факт согласуется с экспериментальными данными для кристаллического DCV4T [23], в котором также доминирует дырочная проводимость. Абсолютные значения подвижности дырок, полученные в ходе нашего расчета для данного материала, согласуются в пределах одного порядка величины с результатами исследования [7], выполненного для аналогичной системы. Однако подвижность дырок значительно ниже, чем наблюдаемая в эксперименте проводимость в кристаллическом DC4VT. Этот факт объясняется различиями структуры молекул материала и его трехмерной упаковки. Экспериментальная плотность кристаллического DC4VT выше, т.е. упаковка молекул значительно более плотная, со значительно большим числом контактов, благоприятствующих межмолекулярному переносу заряда. В работе [7] показано, что проводимость в кристаллической структуре на шесть порядков выше, чем в аморфной, за счет уменьшения энергетического разупорядочивания в системе.
В материале на основе U10–CV2T также доминирует дырочная проводимость: подвижность электронов примерно в 7 раз ниже. Однако абсолютные величины подвижности дырок в этой системе примерно в 20 раз ниже, чем в DCV2T. Это понижение подвижности, по нашему мнению, связано с бóльшим значением энергии реорганизации и меньшими значениями интегралов переноса, а также меньшим количеством контактирующих молекул (пар из списка соседей). Возможно, последний факт объясняется тем, что боковые цепи молекул имеют недостаточную длину для образования тесных контактов.
Изучение траекторий движения в аморфном полимерном материале на основе DCV2T и U10–CV2T показывает, что движение носителя заряда под влиянием приложенного поля в целом происходит линейно. Типичная траектория движения электрона показана на рис. 5а и б. Однако, как видно из этого рисунка, даже в этом случае движение происходит не идеально прямолинейно, а характеризуется значительными по амплитуде случайными движениями, в том числе в противоположных направлениях.
Такая траектория отражает квантовый характер движения носителя заряда, который испытывает квантовые осцилляции при возможности туннелирования между различными сайтами. При совпадении энергий ЛМО (случайно изменяющихся под влиянием тепловых колебаний молекул) заряд может туннелировать даже в направлении, противоположном полю, причем это туннелирование окажется направленным, если обратное туннелирование будет невозможным из-за изменившихся энергий МО и/или увеличившейся под влиянием тепловых колебаний ширины контакта.
Очевидно, что для полномасштабного осуществления “нейросетевого” движения, т.е. движения носителей в зависимости от присутствия на данной молекуле другого заряда, требуется модификация структуры олигомеров. Такая модификация прежде всего касается увеличения длины ответвлений, образования на этих ответвлениях специальных химических групп, обеспечивающих более тесные контакты, и подбора центральной части молекул таким образом, чтобы обеспечить задержку заряда на них. Последнее необходимо для возможности переноса заряда только при нахождении двух или нескольких носителей на “ядре”, т.е. осуществления суммирования сигнала.
Логично ожидать, что для осуществления суммирования сигнала энергия ЛМО центрального фрагмента (“ядра”) в случае движения электрона должна быть низшей в схеме “ядро с ответвлениями”, т.е. низшим должно быть значение энергии низшей свободной МО, а в случае движения дырки – наивысшим, т.е. у “ядра” значение энергии высшей занятой МО должно быть больше, чем у ответвлений. Такой схеме соответствуют многие варианты сборки исследованных молекулярных фрагментов, в том числе вариант P-CV2T, который на данный момент рассматривается нами как наиболее перспективный.
ЗАКЛЮЧЕНИЕ
Для модельных органических материалов на основе производных дициановинилолиготиофенов, хинонов, порфинов, фталоцианина, фитола, рассматриваемых в качестве основы перспективных нейроморфных материалов, методом квантовой химии установлены структурные, энергетические и электронные параметры. Показано, что методы DFT (M062X/cc-pVTZ и M11/cc-pVTZ) хорошо воспроизводят экспериментальные молекулярные структуры, потенциалы ионизации и сродство к электрону изученных молекул. Для двух систем (DCV2T, U10–CV2T) определены параметры их аморфных упаковок и электронные параметры, ответственные за межмолекулярный перенос заряда. В исследованных аморфных материалах дырочная проводимость преобладает над электронной, что согласуется с другими теоретическими и экспериментальными работами. Основываясь на значениях рассчитанных энергий реорганизации, а также на величинах энергий граничных орбиталей и их форме, установлено, что система P–CV2T среди исследованных систем обладает наиболее подходящими свойствами нейроморфного материала, сформулированными в работе. Полученные результаты позволяют указать направления возможной модификации молекул для улучшения свойств, необходимых для создания органических нейроморфных материалов.
Работа выполнена в рамках Программы стратегического академического лидерства ННГУ “Приоритет2030” (проект Н-473-99).
Список литературы
Громов С.П., Чибисов А.К., Алфимов М.В. // Хим. физика. 2021. Т. 40. № 4. С. 9; https://doi.org/10.31857/S0207401X21040099
Fukuzumi S., Honda T., Kojima T. // Coord. Chem. Rev. 2012. V. 256. P. 2488; https://doi.org/10.1016/j.ccr.2012.01.011
Gatty M.G., Kahnt A., Esdaile L.J., Hutin M. // J. Phys. Chem. B. 2015. V. 119. № 24. P. 7598; https://doi.org/10.1021/jp5115064
Илатовский В.А., Птицын Г.А., Комиссаров Г.Г. // Хим. физика. 2008. Т. 27. № 12. С. 52.
Rich P.R. // Farad. Disc. Chem. Soc. 1982. V. 74. P. 349; https://doi.org/10.1039/DC9827400349
Лобанов А.В., Мельников. М.Я. // Хим. физика. 2019. Т. 38. № 7. С. 11; https://doi.org/10.1134/S0207401X19070112
Schrader M., Körner C., Elschner C., Andrienko D. // J. Mat. Chem. 2012. V. 22. P. 22258; https://doi.org/10.1039/C2JM34837C
Koerner C., Ziehlke H., Fitzner R. et al. // Adv. Polym. Sci. 2016. V. 272. P. 51; https://doi.org/10.1007/978-3-319-28338-8_3
Schrader M., Fitzner R., Hein M., Elschner C. // J. Amer. Chem. Soc. 2012. V. 134. P. 6052; https://doi.org/10.1021/ja300851q
Krasilnikov P.M. // Biophys. J. 2014. V. 59. № 1. P. 52; https://doi.org/10.1134/S0006350914010059
Choi M., Shin S., Davidson V.L. // Biochemistry. 2012. V. 51. № 35. P. 6942; https://doi.org/10.1021/bi300817d
Кругляк Ю.А. // ЖФХ. 2014. Т. 88. № 11. С. 1648; https://doi.org/10.7868/S0044453714110144
Painelli A., Girlando A. // J. Chem. Phys. 1986. V. 84. P. 5655; https://doi.org/10.1063/1.449926
Marcus R.A. // Rev. Mod. Phys. 1993. V. 65. № 9. P. 600; https://doi.org/10.1103/RevModPhys.65.599
Troisi A., Orlandi G. // Phys. Rev. Lett. 2006. V. 96. № 8. P. 086601; https://doi.org/10.1103/PhysRevLett.96.086601
Brédas J.-L., Beljonne D., Coropceanu V. et al. // Chem. Rev. 2004. V. 104. № 11. P. 4971; https://doi.org/10.1021/cr040084k
McKechnie S., Booth G.H., Cohen A.J. et al. // J. Chem. Phys. 2015. V. 142. P. 194114; https://doi.org/10.1063/1.4921037
All-atom automatic OPLS-AA topology generator TPPMKTOP; http://erg.biophys.msu.ru/tpp/
Ruhle V., Junghans C., Lukyanov A. et al. // J. Chem. Theory Comput. 2009. V. 5. № 12. P. 3211; https://doi.org/10.1021/ct900369w
Ruhle V., Lukyanov A., May F. et al. // J. Chem. Theory Comput. 2011. V. 7. № 10. P. 3335; https://doi.org/10.1021/ct200388s
May V., Kühn O. Charge and Energy Transfer Dynamics in Molecular Systems. Weinheim, Germany: WILEY-VCH, 2011.
Kirkpatrick. J. // Intern. J. Quant. Chem. 2007. V. 108. № 1. P. 51; https://doi.org/10.1002/qua.21378
Fitzner R., Reinold E., Mishra A. et al. // Adv. Func. Mater. 2011. V. 21. № 5. P. 897; https://doi.org/10.1002/adfm.201001639
Huang X., Nakanishi K., Berova N. // Chirality. 2000. V. 12. № 4. P. 237; https://doi.org/10.1002/(SICI)1520-636X(2000)12:4< 237::AIDCHIR10>3.0.CO;2-6
Ostroverkhova O. // Chem. Rev. 2016. V. 116. № 22. P. 13279; https://doi.org/10.1021/acs.chemrev.6b00127
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика